博文
自动氘气生产新技术
||
尽管重氢(D2)在医疗、核能和化学领域的应用极为广泛,但其在自然界中的丰度极低,这凸显了发明新的重氢生产方法的重要性。我们展示了一种自发的重氢发生器,通过解耦OD-/D+双离子的直接化学反应,利用质子氧化还原反应同时产生相应的电能。这种放热电化学过程在连续运行近85小时内产生了约357毫升的D2,伴随相应的电能输出为每摩尔D2 122千焦。这一实验室级别的自发重氢生产演示为非原生D2的规模化制造提供了一种新的化学方法。
虽然氘是宇宙中第三丰富的元素,但在地球上的自然丰度仅为0.01%。重氢在包括核磁共振、药物开发、医药化学、生化示踪剂、氘化工业化学品合成以及核反应堆中的中子减速剂等多个领域都有广泛应用。由于氘(D2)的核中有一个额外的中子,它在电化学上与轻氢(H2)不同。考虑到其低零点能量、小的离子产品和重水(D2O)显著的脱水能,氘展现出电化学同位素效应(EEI)。唯一非原生D2的生产机制是低温、光催化和电化学过程。有少量证据表明,电解重碱性(NaOD在D2O中)或重酸性(D2SO4在D2O中)水可以产生氘(D2),但这需要将近1.6伏的高电压来启动重氢的电化学生产。然而,这些苛刻条件使得该过程在经济上不可行且大多不方便。
Mondal R, Nayak B, Ottakam Thotiyl M. A Spontaneous Heavy Hydrogen Generator via a Protium Redox. J Phys Chem Lett. 2024 Jun 26:6866-6871. doi: 10.1021/acs.jpclett.4c01399. Epub ahead of print. PMID: 38924762.
与氘合成的吸能过程相反,我们展示了一个在自然天气条件下同时提供电力的自发氘发生器,沿着热力学下坡路径操作。这种自发氘发生器的基础在于通过氢氧化还原作用即时互转换OD-/D+双离子的氧化还原能。一个实验室级别的氘生成原型在环境条件下连续电解近85小时产生了约357毫升的重氢,伴随相应的电能输出为每摩尔D2 122千焦。
这个自发重氢发生器由两个通过Nafion 117膜分隔的半电池组成,如方案1所示。阳极液是重水(D2O)中的钠氘氧化物(NaOD)溶液,使用铂基电催化剂作为阳极电催化剂。阴极液是重水(D2O)中的二氘硫酸(D2SO4)溶液,使用铂基电极作为阴极电催化剂。Nafion 117膜用D2SO4/D2O溶液处理12小时以进行活化。在这种架构设计中,直接酸(pD=0)碱(pD=14)化学被解耦,并且在两个半电池之间将发展出约0.85伏(ΔG°=−83.5 kJ/mol)的内在电动势(计算S1)。然而,由于OD-/D+双离子的再结合反应是非氧化还原性的,它不能直接作为电动力收获。为了使这种直接互转换成为可能,采用了氢氧化还原反应,因为酸碱反应正是碱性电解质和酸性电解质中氢氧化还原之间的差异(方程1和2)。这表明在碱性阳极液中消耗了氢,同时在酸性阴极液中伴随着氢的产生。因此,总的反应化学不涉及任何净消耗氢气燃料(方程3)。通过用D2SO4代替H2SO4替换酸性半电池,这种化学应该以牺牲阳极半电池中的氢气燃料为代价,在阴极产生D2而不是H2,方程4至6。
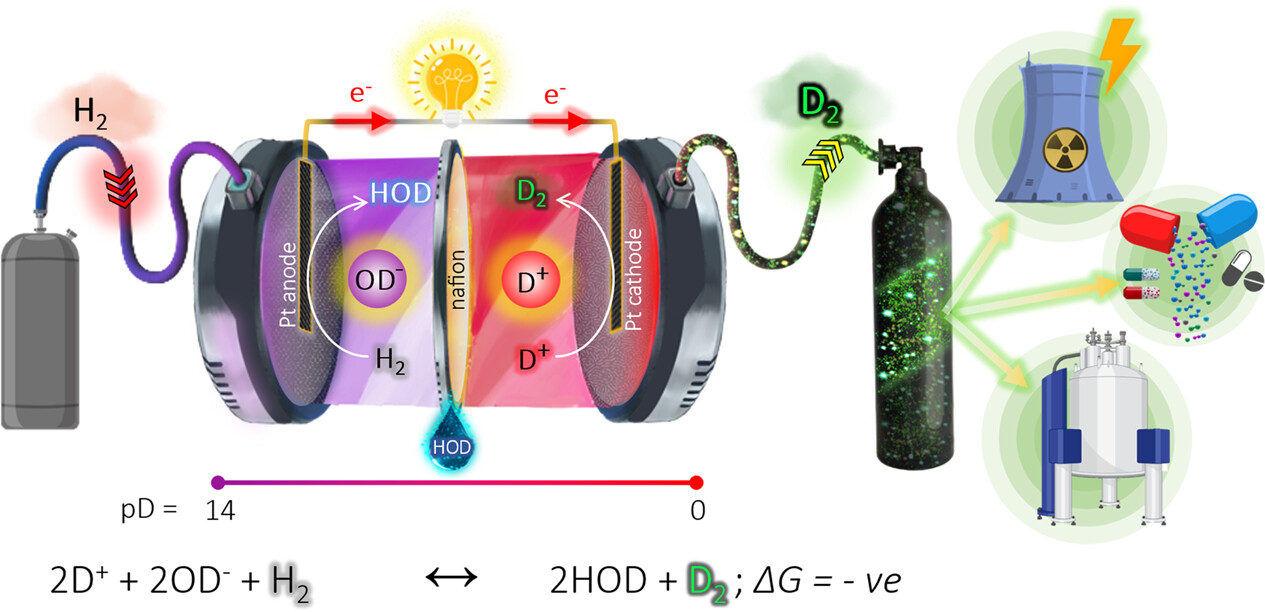
Scheme 1. Schematics of Spontaneous Electrochemical Deuterium Generator under Ambient Conditions
In the case of the hydrogen system,
Anodic half‐cell reaction:2H2O+2e−↔H2+2OH− (1)
Cathodic half‐cell reaction:2H++2e−↔H2 (2)
Overall cell reaction:2H++2OH−↔2H2O (3)
In the case of the deuterium system,
Anodic half‐cell reaction:2HOD+2e−↔H2+2OD− (4)
Cathodic half‐cell reaction:2D++2e−↔D2 (5)
Overall cell reaction:2D++2OD−+H2↔2HOD+D2 (6)
为了检验其可行性,我们在酸性和碱性介质中探测了氘和氢的氧化还原反应。在循环伏安图中,阳极峰对应于氘/氢氧化反应(DOR/HOR),而阴极电流则归因于氘/氢析出反应(DER/HER)。在氘化硫酸溶液(D2SO4)中,与H2SO4相比,氘析出反应向负方向偏移,相应的碱性溶液也显示出类似的偏移,图1a,b和表S1。这归因于氘较低的零点能结合重水的高脱水能引起的电化学同位素效应(EEI)。对于D2O,由于其较低的零点能导致较高的活化能,从而使得氘析出反应具有相对较高的过电势。(11)D2O较小的离子产物也意味着溶液中D+离子浓度较低,从而导致氘析出反应速率降低。(11,13)D和H之间零点能(ZPE)的差异导致了D2O和H2O之间的溶剂化或水合能的差异。由于从H到D的质量增加,D系统的核-电子振动变弱,导致D2O的情况下溶剂化能更高。(11)D2O较高的脱水能也使得氘析出反应(DER)与H2O中的氢析出反应(HER)相比,电化学上较不利。(11,14)由于D系统的熵变比H系统更正,因此前者从环境中获取的能量将比后者更高(计算S1和S2)。这导致D系统的热力学效率略高于H系统(分别为1.45和1.43)(计算S2)。为了研究动力学行为,我们推导了酸性和碱性介质中氢和重氢析出反应(HER/DER)的塔菲尔斜率值。观察到在两种介质中,HER的塔菲尔斜率都比DER低,图S1,导致HER的反应动力学比DER更快。这可能归因于D2O相对较低的零点能及其与基于H的系统相比较小的离子产物。(10−14)总之,这些特性导致D2O在其活性上与H2O相比有明显的电化学差异。与此一致的是,观察到碱性介质中的氢氧化还原(pD = 14, 1 M NaOD)与酸性介质中的氘氧化还原(pD = 0, 0.5 M D2SO4)之间存在约0.85 V的正电位差(计算S2),图1c和图S2。单电极电位图1d表明,在设备配置中,供氢的碱性半电池表现出比氘析出的酸性半电池更负的电位,从而导致整体正电池电位,图1d。
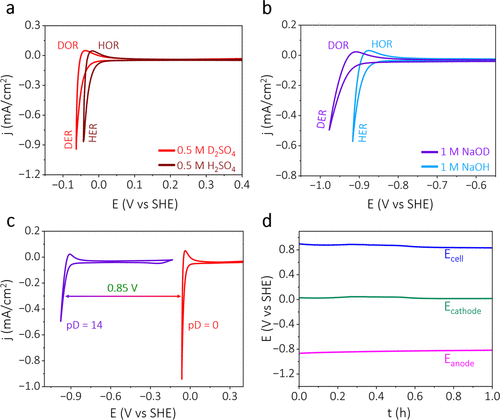
图1. pH依赖的氢/氘氧化还原反应。(a) 酸性电解质中的氢氧化还原(酒红色迹线)和重酸性电解质中的氘氧化还原(红色迹线)。(b) 碱性电解质中的氢氧化还原(天蓝色迹线)和重碱性电解质中的氘氧化还原(紫色迹线)。(c) 根据电解质之间双离子浓度梯度(pD梯度)可获得的可用电位差。(d) 两电极装置半电池的单电极电位,其中pD梯度为14(阳极)和0(阴极)。
因此,重碱性(NaOD)阳极半电池在供给氢气(H2)时应该发生氧化并释放电子(方程4)。在获得这些电子后,相应的氘析出反应(方程5)应该在重酸性电解质(D2SO4)的阴极半电池发生。由于在输送电能期间以轻氢(H2)为代价产生重氢(D2)(方程6),该装置被命名为自发重氢发生器。这种氘发生器的极化曲线显示出在电流密度接近115 mA/cm2时的峰值功率密度约为45 mW/cm2(图2a中的紫色迹线)。当D2析出被H2析出替代时,性能比较几乎相同(图2a中的橙色迹线)。应该注意的是,在没有氢作为阳极进料的情况下,未观察到明显的功率密度,这表明H2是这种氘发生器的燃料,图S3。在恒定电流密度40 mA/cm2下的恒电流极化显示出自发氘析出的稳定电压输出(图2b中的紫色迹线)。随着时间的推移,由于各自半电池中H+/D+和OH–/OD–双离子梯度的持续下降,观察到电位输出逐渐下降,计算S3。当氘析出被氢析出替代时,观察到类似的行为,图2b中的橙色迹线。方程6表明,这种电池的寿命依赖于两个半电池之间的OD–/D+双离子浓度梯度。在电化学氘析出前后估计了双离子浓度,发现在长期恒流测试后两者都显著减少,图2c,d。进行恒流测试后,确定了剩余的双离子浓度(图2c,d),并且它们的消耗量与恒流测试期间通过的电荷量相当。当从装置中抽取电流近5小时(图2b)后,剩余的电解质仅含约15%的双离子(H+/D+和OH–/OD–),图S4。当不抽取电流时,装置的双离子浓度在长达50小时的时间内波动最小,图S5。综合这些结果表明,在自发重氢生成过程中(如方程6所示),双离子是通过电化学途径消耗的。
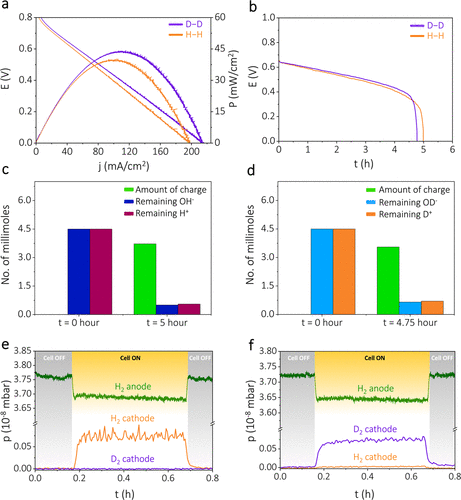
图2. 从轻氢自发生成重氢。(a) 双室装置的极化曲线,具有NaOH-H2SO4(橙色迹线)和NaOD-D2SO4(紫色迹线)双电解质系统。(b) 两种装置在恒定电流密度40 mA/cm2下的恒流极化。在恒流极化期间,通过的电荷与阴极电解液中H+/D+浓度和阳极电解液中OH–/OD–浓度变化的相关性,对于(c) NaOH-H2SO4电解质和(d) NaOD-D2SO4电解质。装置的现场电化学质谱,具有(e) NaOH-H2SO4双电解质和(f) NaOD-D2SO4双电解质。
为了研究该装置的工作机制,对具有H+/OH–双离子梯度的NaOH-H2SO4进行了现场电化学质谱分析,图2e。在现场电化学质谱中,注意到阳极半电池中氢气分压的降低(图2e中的绿色迹线所示),伴随着电池运行期间阴极半电池中氢气分压的同时增加(图2e中的橙色迹线所示)。这表明在碱性阳极消耗了H2,并在阴极半电池中发生了氢气析出,从而证实了方程4。然而,在具有D+/OD–双离子梯度的NaOD-D2SO4系统中,观察到当电池开启时,阳极半电池消耗了H2(图2f中的绿色迹线)。同时,在阴极半电池产生了D2(图2f中的紫色迹线)。这表明以阳极处的H2为代价析出了D2,证实了方程5。值得注意的是,也析出了少量的HD(其量如下所述进行了量化),这归因于重水溶剂中作为杂质存在的微量水。
为了量化析出的氘气的数量和法拉第效率,我们将通过的电荷与产生的D2、HD和H2的数量进行了关联。对氘发生装置的阴极半电池进行了现场电化学质谱分析,图3a。它显示出D2的法拉第效率接近92%,HD为6.5%,H2为1.5%(图3b,计算S4)。在不同电流密度下的速率能力已经进行测试,如图S6所示,证明了该装置能够维持相对较高的速率。通过水置换技术量化了包括D2、H-D和H2在内的总气体析出量,这与通过的总电荷量线性相关,图S7。所有三种电流密度下整个气体析出的累积法拉第效率接近100%,图S8。进一步,通过各种电流额定值监测氘析出期间的电势输出,采用计时电位阶梯法。这种行为与阴极半电池的相应现场电化学质谱相关联,图S9。显而易见,随着电流额定值的增加,电势随之下降,同时D2析出率相应增加。有趣的是,当增加电流额定值时,该装置迅速返回到几乎原始的电压平台,D2析出率呈现相似趋势,图S9。这种氘发生装置的耐用性已经过检验,显示出在约85小时的持续电解下,它可以一致地产生D2,恒定电流密度为10 mA/cm2,图3c。在氘析出期间得出的电能输出几乎为每摩尔D2 122.4 kJ,图3d,计算S5。
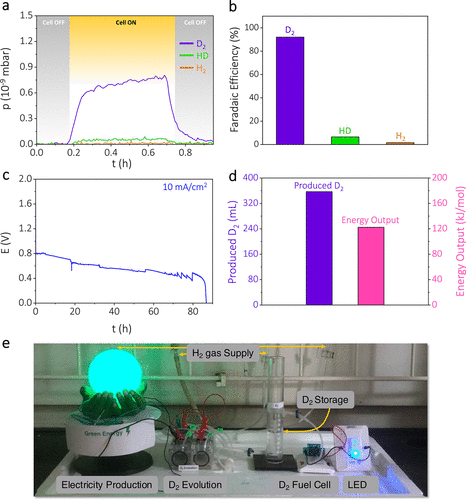
图3. 自发重氢发生器的实验室级演示。(a) 具有NaOD-D2SO4双电解质系统的电化学氘发生器阴极半电池的现场电化学质谱。(b) 电化学氘析出的法拉第效率。(c) 在10 mA/cm2的电流额定值下,氘发生装置的长期耐用性测试近85小时。(d) 自发重氢发生器的氘析出量和电能输出的量化。(e) 照片展示了自发电化学氘发生器直接作为D2空气燃料电池的D2储存器的使用。
在长期重氢析出过程中,通过现场电化学质谱在不同时间间隔测定了阴极排气中重氢的比例,图S10。随着时间的推移,D/H比率随着D2量的减少而降低,HD的量显著增加(图S11和计算S6)。这可能是由于阳极半电池中形成的HOD(通过扩散和渗透流)运输到阴极半电池,并随后参与阴极半电池反应。随着装置的连续运行,这种运输将增加,因为越来越多的HOD在阳极电解液中积累(式4)。这可能解释了在长期重氢生成过程中HD含量的增加(或D/H比率的降低)(图3c和图S11)。然而,在长期实验开始时检测到了少量的HD(近6%),这可能是由于电解质中存在工作气氛中的水分轻微污染所致。累计析出的氘气量接近357 mL。为了展示电化学D2发生器的实用性,我们已经制造了一个实验室级别的自发D2发生器,并将其与D2空气燃料电池串联连接(图3e和支持信息视频S1)。这一演示验证了自发电化学重氢发生器可以有效地作为D2空气燃料电池的D2储存器,利用其在重氢生成期间产生电力的能力。简而言之,通过利用D+/OD–双离子梯度能量,在环境天气条件下以轻氢为代价同时获得电力和重氢。
我们已经展示了在环境天气条件下从轻氢生成重氢,伴随着电能输出,通过操纵OD–/D+双离子梯度能量实现。这种自发重氢发生器通过离子导电膜解耦直接的OD–和D+化学反应,以热力学下降的方式工作。实验室规模的重氢发生器原型可以在近115 mA/cm2的电流密度下提供近45 mW/cm2的峰值功率密度,同时在连续运行85小时期间生成近357 mL的重氢,每摩尔D2的总能量输出为122 kJ。值得注意的是,设备的连续运行会导致阳极电解液中含氕物种的积累,这最终应导致同位素选择性的降低。因此,为了长期保持重氢的纯度,需要定期补充电解质。尽管如此,这里采用的方法呈现了一种独特的化学过程,用于收获OD–/D+双离子的能量作为电驱动力,尽管OD–和D+组合是一种非氧化还原置换反应。这种置换反应的能量是使用氕氧化还原作为电驱动力收获的,因为在解耦的OD–/D+配置中阳极和阴极半电池反应之间的差异对应于在电化学途径中发生的非氧化还原反应。
https://blog.sciencenet.cn/blog-41174-1439955.html
上一篇:肿瘤微生物组学《自然》重要论文撤稿
下一篇:镁合金血管支架通过释放氢气产生脑保护作用