博文
果糖和高尿酸血症  精选
精选
|
果糖和高尿酸血症
随着高尿酸血症的广泛流行,它已经成为人类公共健康的一个严重威胁。越来越多的证据表明,饮食中的果糖与高尿酸血症有着密切的关系,但果糖摄入在高尿酸血症中的作用仍然不明确。高尿酸血症的特点是尿酸盐晶体的过量产生和沉积。果糖的代谢导致血清尿酸浓度增加。在这篇综述中,我们描述了全球果糖消费的最新情况以及高尿酸血症的流行病学,并总结了研究果糖摄入与高尿酸血症风险之间关系的研究进展。本综述强调了果糖在肝脏、小肠和肾脏中的代谢过程。此外,我们还讨论了果糖代谢的分子机制,以揭示其背后的机理。同时,我们详细阐述了果糖代谢对高尿酸血症的影响,以深入理解由果糖摄入引起的高尿酸血症的发病机制。果糖消费与增加的高尿酸血症风险密切相关。为了理解果糖摄入在高尿酸血症发展中的作用,更多的前瞻性研究是不可避免的。
Recent advances in fructose intake and risk of hyperuricemia - ScienceDirect
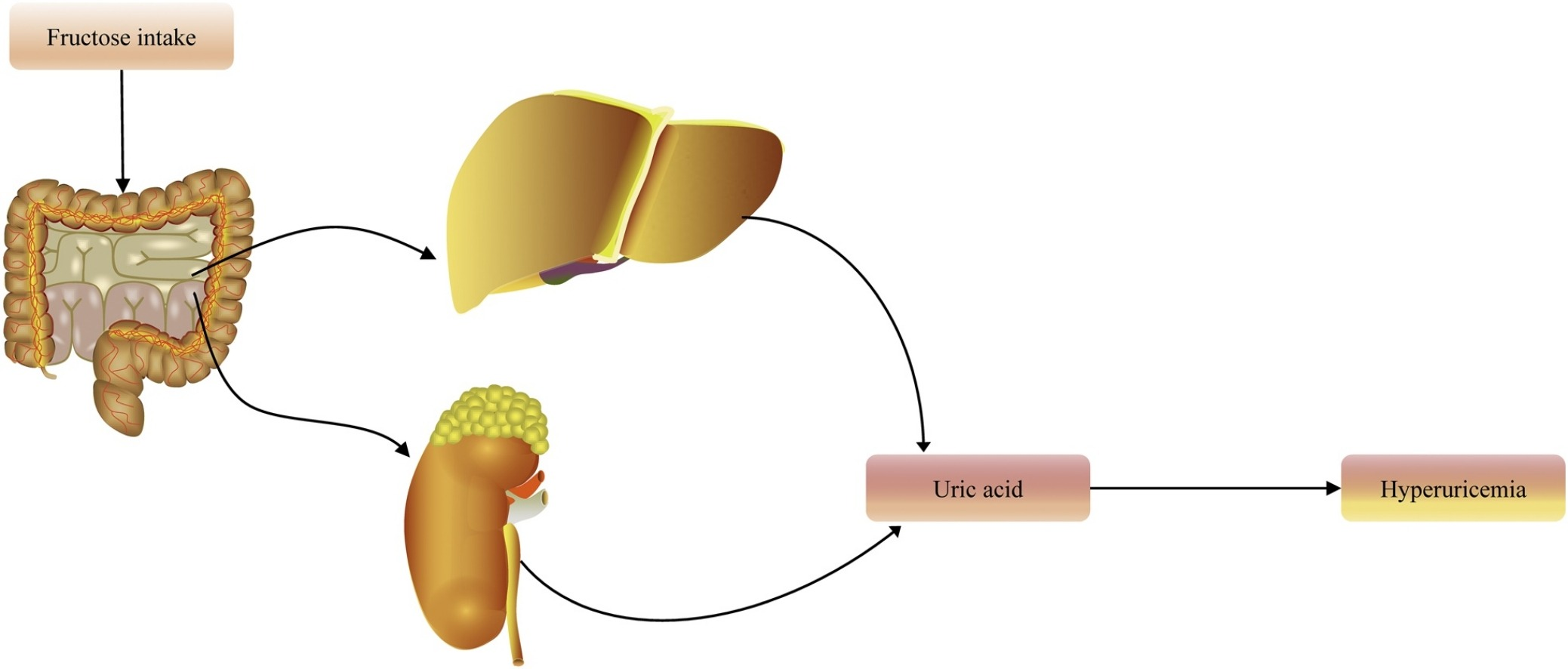
随着经济的快速发展和生活方式的转变,传统的以蔬菜和碳水化合物为主食的饮食模式已转向肉类、乳制品和其他富含嘌呤的食物,这与高尿酸血症的普及密切相关。高尿酸血症已逐渐发展成为一个全球性的公共健康问题。
过去几十年来,血清尿酸水平的上升和高尿酸血症的流行已经引起了越来越多的关注。在经济发达国家,高尿酸血症的发病率尤其高。相比之下,发展中国家的高尿酸血症发病率似乎较低。根据国家健康与营养调查的结论,大约21%的美国成年人患有高尿酸血症。高尿酸血症是由于血清尿酸过多而产生的,这可能是因为过量产生或排泄不足导致的,可以引发痛风。由于尿酸过量产生的高尿酸血症与各种疾病有关,如骨髓增生性疾病、肿瘤溶解综合症,占不到10%。而由于尿酸排泄不足引起的高尿酸血症占90%以上,与肾功能障碍有关。此外,高尿酸血症被认为是代谢综合征、慢性肾病、心血管疾病和糖尿病的标志。各种因素,如过量摄入果糖和滥用酒精,都会导致高尿酸血症的发生。
尿酸主要在肝脏、肌肉和脂肪组织中产生,并主要通过肾脏排泄。尿酸水平的升高与糖尿病、心脏病和肾病有关。此外,由于过量产生或排泄减少而导致的尿酸水平升高也与嘌呤降解增加、代谢性酸中毒和肾功能不全有关。一旦尿酸的产生和排泄之间的不平衡导致高尿酸血症,过多的尿酸就会在关节中积累。它会引起尿酸晶体的产生,导致组织和关节的疼痛和炎症。降低血清尿酸水平可以使尿酸晶体逐渐在血液中溶解,从而使溶解的尿酸从体内排出。同时,低血清尿酸水平有利于抑制新晶体的形成。因此,黄嘌呤氧化酶抑制剂通过与黄嘌呤氧化酶相互作用来抑制尿酸的产生,而尿酸排泄剂则通过减少尿酸的重吸收来提高尿酸的排泄。
多种饮食因素可以通过提高嘌呤的产生和减少尿酸盐的排泄来促进高尿酸血症的发生,包括高酒精摄入量和摄入富含嘌呤的食物,如海鲜或红肉。果糖是一种广受欢迎的食品成分,近年来果糖的消费逐渐增加。果糖的摄入可以通过刺激腺苷核苷酸的分解代谢来促使高尿酸血症的发生。此外,来自工业甜饮料的果糖摄入量增加与大型人群样本中颈动脉-股动脉脉搏波速度的增加有关。此外,大量证据指出,高果糖摄入与各种疾病的风险增加有关,如痛风、高尿酸血症、肥胖、高血压、2型糖尿病、心血管疾病和非酒精性脂肪肝病。尽管有一些关于果糖消费与高尿酸血症之间关系的研究存在冲突,但不可否认的是,果糖摄入可能与高尿酸血症的风险有关。因此,加深对果糖代谢过程及其潜在分子机制的理解至关重要。特别是,对果糖与高尿酸血症之间关系的新见解可能会为高尿酸血症提供更精确的治疗。
鉴于高尿酸血症的广泛流行,澄清和阐述可能导致高尿酸血症风险的果糖摄入变得至关重要。因此,评估全球果糖消费情况和高尿酸血症的流行病学是必要的。
2.1 全球果糖消费情况
在过去的几十年里,有确凿的证据表明,由于食物消费模式向富含高热量、含糖饮料和含有高果糖玉米糖浆或蔗糖的加工食品转变,果糖消费显著增加。根据美国农业部(USDA)的记录,从1977年到2004年,平均果糖摄入量从每天37克增加到49克。非美国人群也呈现相同的趋势。根据国际糖组织的报告,1986-2007年间,人均糖消费量从每天56克增加到65克。在荷兰国家食品消费调查2007-2010中,7至69岁荷兰人口的中位果糖摄入量为每天46克,略低于美国人口。此外,应用一个全面的淀粉和糖成分数据库来估计日本人口的淀粉和糖摄入量,结果表明由于饮食差异,日本人口的淀粉摄入量与美国人口相比较低。
果糖摄入量的增加主要是由于含糖饮料的消费增加,包括能量饮料、果汁和软饮料,这些是当前饮食中添加糖的主要来源。尽管西欧、澳大利亚和北美的含糖饮料消费逐渐放缓,但在欠发达地区的消费却在稳步加速。此外,美国的糖消费量超过了推荐摄入量,尽管在特定国家的糖消费正在减少。总之,果糖消费的分布和流行确实存在地区差异。
2.2 高尿酸血症的流行病学
世界上已有许多关于高尿酸血症流行病学的研究,这意味着高尿酸血症引起了越来越多的关注。在2015-2016年,美国成年人高尿酸血症的总体患病率高达14.6%,相当于约有3250万受影响个体。尽管年龄调整后的高尿酸血症患病率从2007-08年的3250万略有下降到2015-16年的3210万,但受影响个体的数量是相同的。一项提供类似结果的全国代表性研究表明,尽管高尿酸血症在2007年至2016年间保持稳定,但仍然居高不下。在2015-2016年,患有高尿酸血症的女性和男性人数分别达到2440万和2280万。此外,发现老年患者尤其是80岁以上个体的高尿酸血症患病率更高,最高患病率为27.8%。一项包括14187名参与者的横断面研究显示,这些人可以代表1988-1994年间美国平民非机构化人口,超重(人群归因风险=44%)、不遵循DASH风格饮食(人群归因风险=9%)、饮酒(人群归因风险=8%)和使用利尿剂(人群归因风险=14%)与高尿酸血症有很大关系。根据2016年4月至2017年3月间的日本健康保险索赔和医疗检查数据,无症状高尿酸血症和高尿酸血症在参与横断面研究的2531383人中分别占2.6%和13.4%。有趣的是,在韩国中年女性中,普遍的高尿酸血症从更年期晚期过渡阶段开始急剧增加。
作为一个地域广阔、人口众多的发展中国家,中国在不同地区的高尿酸血症流行病学上存在一些差异。基于2014-2018年中国成人人口的医疗数据,推测2018年约有2.71亿中国成人可能患有高尿酸血症。一项包括2014年1月至2015年12月来自中国东部9225名居民的区域调查显示,高尿酸血症的患病率为11.3%,其中男性为10%,女性为12%。此外,城市受试者高尿酸血症的患病率高于农村受试者(分别为12.9%和10.8%)。荟萃分析表明,中国农村地区的高尿酸血症相对较高,患病率高达11.7%。简而言之,高尿酸血症的广泛流行已严重危害了人类健康。
2.3 果糖摄入与高尿酸血症的风险
越来越多的证据表明,添加糖,尤其是果糖,对代谢有不利影响,导致痛风、高尿酸血症、肥胖、高血压、2型糖尿病、心血管疾病和非酒精性脂肪肝病。然而,高膳食果糖摄入与高尿酸血症风险增加之间的关系仍然存在争议。
根据1999-2004年全国健康和营养调查(NHANES)数据库和逻辑回归模型,未发现果糖摄入与高尿酸血症风险之间的显著相关性。另一项荟萃分析显示,含糖饮料和果汁的摄入与痛风风险之间存在不利关联,但未发现与水果摄入的关联。更多研究表明,高果糖摄入可以导致血液中尿酸浓度升高,而由嘌呤代谢紊乱引起的高尿酸血症是血液中尿酸水平过高。一项涉及125,299名参与者的荟萃分析显示,高果糖消费与痛风风险增加相对应。同样,一项包括244只不同果糖喂养的大鼠的荟萃分析表明,高尿酸血症与高果糖摄入之间存在显著关系。另一项系统综述和荟萃分析显示,成人中含糖饮料消费与痛风和高尿酸血症风险增加之间存在实质性关联。基于巴西成人健康纵向研究(ELSA-Brasil)基线数据(2008-2010),一项横断面调查显示,高果糖摄入与高尿酸血症之间存在显著相关性,中度和高果糖摄入与高尿酸血症之间存在实质性相关性。总之,大多数研究表明,高果糖摄入可能导致高尿酸血症。
3. 果糖的代谢
3.1 果糖在肝脏中的代谢
葡萄糖代谢在体内广泛进行,而果糖的代谢主要在肝脏中进行。摄入的果糖在肝脏中主要用于补充肝糖原并生成甘油三酯。果糖代谢的特殊性在于它在代谢过程中减少ATP和细胞内磷酸盐(图1)。

图1. 果糖在肝脏中的代谢。果糖摄入导致AMP降解为IMP,并刺激尿酸的产生。Fructose-1P, 果糖-1-磷酸酯;DHAP, 二羟丙酮-P。
不仅己糖激酶,酮己糖激酶也能介导果糖的代谢[53,54]。果糖主要由酮己糖激酶代谢,该酶被称为果糖激酶C(KHK-C)或KHK,快速生成的果糖-1-磷酸酯导致ATP和磷酸化水平下降。细胞内磷酸盐的减少增强了AMP脱氨酶的活性,导致AMP转化为IMP,并刺激尿酸的产生,加速了肌苷单磷酸(IMP)和尿酸的产生。随后,果糖-1-磷酸酯可以通过果糖-1-磷酸酯醛缩酶(醛缩酶B)和各种酶代谢,产生乳酸、糖原、葡萄糖和甘油三酯[25,54,55]。
根据果糖代谢的组织不同,果糖表现出不同的生理功能。当摄入少量葡萄糖和果糖时,它们可以通过肝脏和小肠迅速清除,几乎不会有果糖排入尿液中。当过量的果糖和葡萄糖诱导更多的果糖和葡萄糖摄入时,会导致野生型小鼠出现代谢综合症和脂肪肝,这导致通过KHK的果糖代谢增加。KHK缺陷型小鼠缺乏KHK可以降低葡萄糖和果糖的摄入,并抑制果糖代谢。小肠中的KHK调节和控制总体热量和糖分的摄入。然而,持续或过量的果糖暴露可导致肠道果糖清除率下降,并有助于将更多果糖输送到肝脏,从而导致代谢综合症、脂肪积累和脂肪肝。相反,尽管阻断肝脏中的KHK不能减少果糖摄入,但它可以完全抑制通过KHK的果糖代谢,并促进通过己糖激酶的果糖代谢或将果糖排入尿液中。肝特异性果糖激酶敲除小鼠中由糖诱导的代谢综合症表明,肝脏中果糖的过度分解促进了多种代谢综合症[54]。果糖激酶C是果糖代谢中的关键酶,可以触发ATP耗尽、核苷酸转换、促进尿酸形成,最终导致脂肪积累。此外,摄入果糖会影响肠道通透性和微生物群,从而增加NAFLD和NASH的风险[48,56,57]。
果糖和葡萄糖补充增强了ChREBP-β的表达。果糖补充提高了SREBP1c的表达和脂肪酸合成,导致肝脏胰岛素信号传导受到抑制。相反,葡萄糖有助于ChREBP的表达和甘油三酯的合成,导致肝脏胰岛素信号传导激活。这意味着葡萄糖补充和果糖补充对高脂饮食小鼠的肝脏代谢产生了显著影响。此外,与葡萄糖补充的高脂饮食小鼠相比,果糖补充的高脂饮食小鼠更有可能发展为葡萄糖不耐症、肝肿大和肥胖。与葡萄糖补充相比,果糖补充增加了丙二酰辅酶A的水平,并有助于ACADL和CPT1a的乙酰化,这与脂肪酸氧化途径密切相关。然而,葡萄糖补充导致HADA/HADB的乙酰化增强,积极参与脂肪酸氧化。在高脂饮食小鼠中,果糖补充不仅可以增加脂肪酸合成,还可以抑制线粒体代谢,这可能导致肥胖[[58], [59], [60]]。
ChREBP作为一种转录激活因子,在糖酵解和脂肪生成中扮演着至关重要的角色。果糖补充增加了肝脏中的己糖磷酸盐水平。它促进了ChREBP的激活,进一步导致了小鼠的高甘油三酯血症、高胰岛素血症、肝脂肪变性和葡萄糖不耐受。果糖激活的葡萄糖-6-磷酸酶是肝脏中产生葡萄糖和葡萄糖-6-磷酸的决定性因素。转录因子叉头框O1a(FOXO1A)可以正向刺激G6pc的表达,而胰岛素信号传导可以介导FOXO1A的抑制。在FOXO1A缺陷的情况下,果糖激活了ChREBP。它刺激了G6PC的表达,表明果糖诱导的ChREBP和G6PC的激活超过了胰岛素的抑制效果。这增强了肝脏中的葡萄糖产生并导致胰岛素抵抗[59,61,62]。
肝脏摄入果糖通过增强胰岛素分泌和刺激脂肪生成基因表达来促进新的脂肪生成。此外,肠道微生物将摄入的果糖转化为乙酸,这一过程产生了独立于ATP柠檬酸裂解酶(ACLY)的脂肪生成乙酰辅酶A[63]。另外,作为果糖代谢必需酶的醛缩酶B(ALDOB)通过GATA6在转移性肝细胞中上调。而且,抑制ALDOB或GATA6会抑制肝脏转移。同样,减少果糖摄入也会限制肝脏转移[64]。
3.2 小肠中的果糖代谢
普遍认为,大部分饮食中的果糖由肝脏代谢[24,65,66]。然而,最近研究人员使用同位素追踪技术发现,实际上是小肠而不是肝脏,在小鼠中将大部分饮食果糖代谢成葡萄糖和有机酸[67,68]。当摄入适量的果糖时,小肠将代谢大约90%的果糖。只有当果糖摄入量过多时,小肠清除果糖的能力才会不堪重负,肝脏主要代谢过量的果糖。此外,与禁食状态相比,在进食状态下,小鼠摄入果糖会导致小肠中代谢增加。进一步研究表明,空腹时大量摄入果糖导致小肠中果糖代谢减少,更多果糖溢出到肝脏[67,69]。当小肠摄入果糖时,大部分被磷酸化并转化为有机酸和葡萄糖。一部分未代谢的果糖通过小肠到达肝脏,这是由于高剂量果糖超过了小肠对果糖的吸收和清除率[70,71]。
最近的研究表明,小肠也可以通过果糖分解和糖异生酶像肝脏一样代谢摄入的果糖,这表明小肠在果糖代谢中起着重要作用。此外,小肠中的果糖分解和糖异生酶由葡萄糖转运蛋白5(GLUT5)和果糖激酶调节和控制[72]。如图2所示,一旦果糖通过GLUT5穿过肠上皮细胞,它就被果糖激酶磷酸化并转化为果糖-1-磷酸,这一过程缺乏反馈抑制。
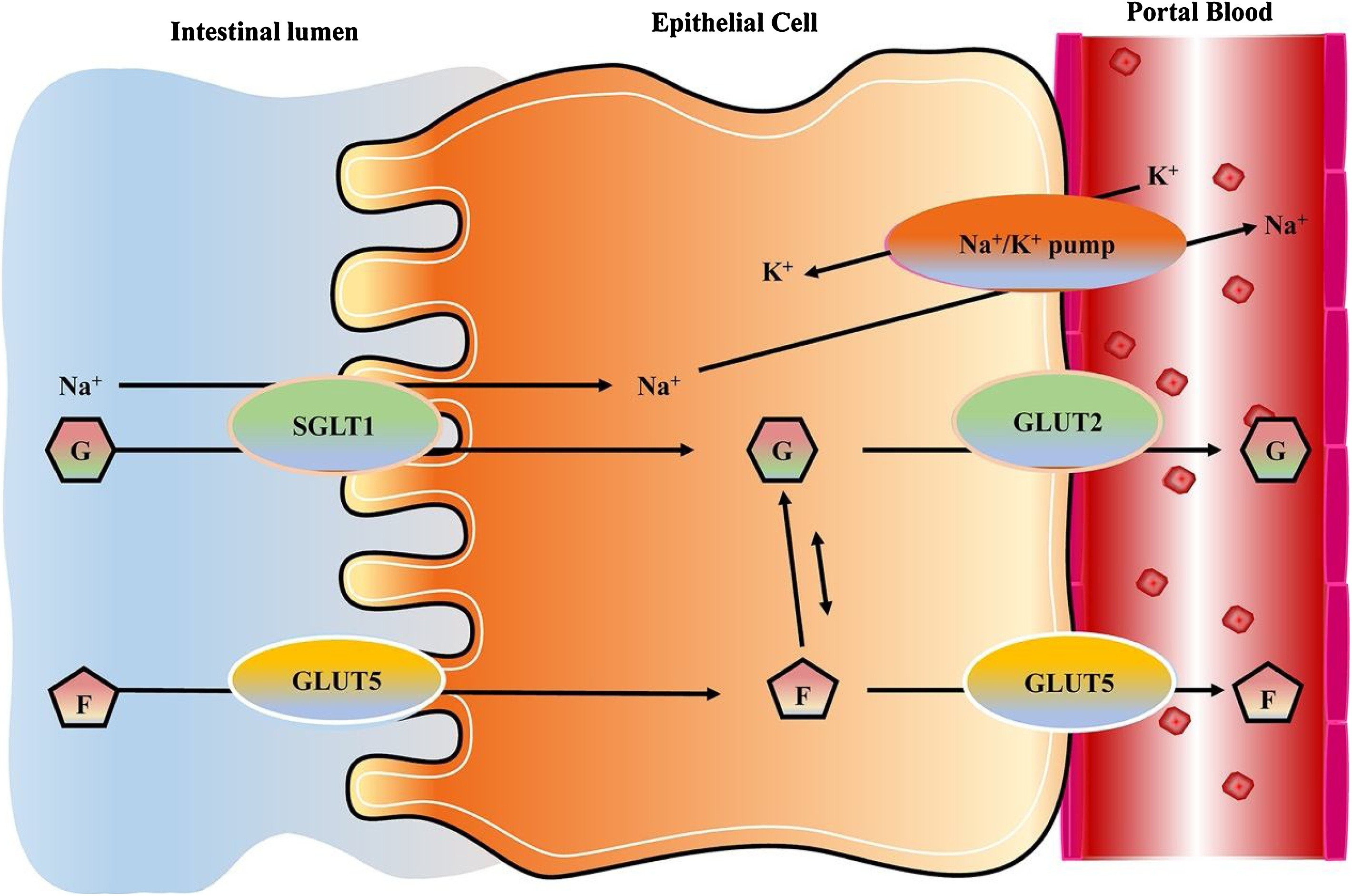
图2. 小肠中的果糖代谢。果糖通过GLUT5穿过肠上皮细胞。G, 葡萄糖;F, 果糖;SGLT1, 钠依赖型葡萄糖共转运蛋白1;GLUT2, 葡萄糖转运蛋白2;GLUT5, 葡萄糖转运蛋白5。
GLUT5是GLUT家族的一个成员,主要负责将果糖吸收到肠细胞的顶膜中,不能转运半乳糖或葡萄糖[65,73]。在小鼠基因模型实验中已经发现,GLUT5有助于果糖通过顶膜的吸收。果糖水平可以调节肠道GLUT5的表达水平,并控制肠道中的果糖转运速率,这与KHK介导的代谢、ChREBP、TXNIP和皮质酮密切相关。过量的果糖摄入可能会降低肠道中的Pi和Ca2+转运,从而可能对骨骼健康产生潜在的不利影响。当肠道果糖吸收改变时,管腔内果糖的浓度也会改变,这会影响肠道微生物的生态[74]。GLUT5和GLUT2介导的果糖对肠道电解质转运、肝脏尿酸代谢以及肾脏和心肌细胞功能的影响可能在果糖引起的高血压中发挥重要作用。此外,GLUT2不仅可以转运果糖,还可以在肠细胞的基底侧部分将半乳糖和葡萄糖转运到血管中[65]。而且,GLUT2可能部分负责通过促进过量果糖摄入来加速非酒精性脂肪肝疾病的进展[75]。
人们普遍认为,尿酸和果糖可以调节小肠中果糖激酶的表达。果糖激酶介导的果糖磷酸化以及促进果糖吸收并将其转化为其他代谢产物是小肠中果糖代谢的起点。此外,其过度表达会诱导果糖的清除并抑制脂质生成。同样,一旦摄入的果糖量超过小肠的代谢能力,果糖可以诱导脂质生成并导致代谢综合症[55,76]。缺氧诱导因子可以调节果糖激酶水平,这也解释了缺氧刺激果糖激酶,而果糖可以在缺氧条件下介导厌氧代谢,从而延长生存时间[77]。此外,高剂量果糖的摄入可以引起肝脏新的脂肪生成,这在肝脏中大多独立于ATP柠檬酸裂解酶(ACLY),并且大部分依赖于肠道微生物将果糖代谢成乙酸[63,66]。
3.3. 肾脏中的果糖代谢
研究表明,静脉注射果糖会导致肾脏和肝脏吸收己糖,伴随着肾脏和肝脏中葡萄糖的产生以及肾脏中乳酸的产生,表明肾脏对于果糖的处理至关重要,且乳酸和葡萄糖是肾脏中果糖代谢的最终产物[78]。
近曲小管细胞使用重吸收的果糖作为糖异生的底物,从而维持血清葡萄糖浓度的平衡。在过量摄入果糖的情况下,果糖的代谢受到限制,导致细胞内能量耗尽、尿酸产生、肾脏纤维化、线粒体氧化应激和炎症。在糖尿病、肾脏疾病和心脏肥大等病理情况下,果糖可以内源性产生,扰乱内源性果糖的代谢可以延缓肾脏损伤的进展。在标准饲养模式下,大鼠肾脏可以产生约26%的血清葡萄糖。同时,在饥饿大鼠的肾脏中发现了46%的葡萄糖释放增加。糖异生可能归因于肾脏中的底物,包括谷氨酰胺、丙酮酸、乳酸、丙氨酸和果糖。
果糖代谢不仅发生在近端直小管,也发生在近端曲小管(图3)。在近端直小管中,葡萄糖转运蛋白5介导尿液中果糖的吸收,随后通过果糖激酶在整个胞浆中代谢。此外,近端曲小管也发现了果糖激酶和醛缩酶B的表达,其中醛缩酶B的缺失导致果糖1-磷酸的积累。在生理条件下,果糖主要在近端肾小管上皮细胞中代谢并转化为葡萄糖。然而,连续摄入不合理量的果糖可能导致其分解,造成大量ATP消耗和炎症,从而导致肾小管损伤[49,79]。
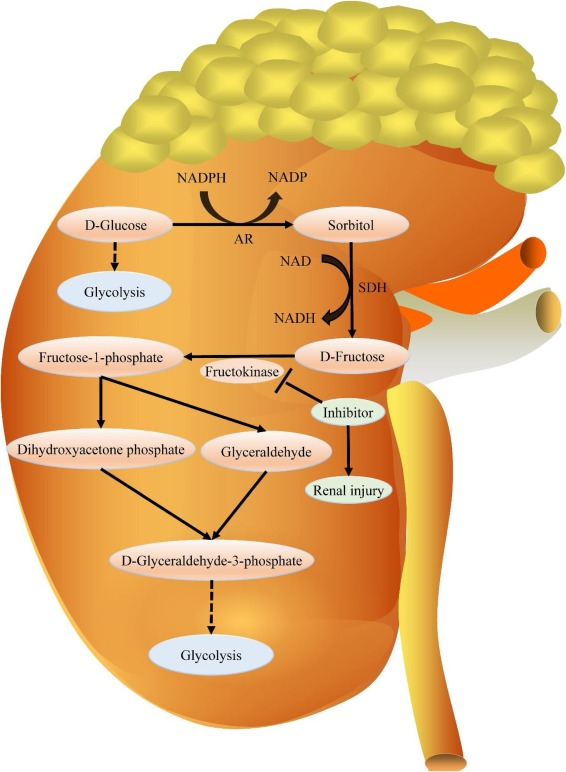
Fig. 3. 果糖代谢发生在近端直小管和近端曲小管。AR, 醛糖还原酶;SDH, 山梨醇脱氢酶。
多元醇通路的激活被认为是内源性产生果糖的主要机制。在多元醇通路中,葡萄糖通过两步转化为果糖,其中醛糖还原酶介导葡萄糖转化为山梨醇的过程。随后,山梨醇脱氢酶介导山梨醇氧化为果糖的过程。值得注意的是,通过肾脏中的果糖新生和糖异生获得的内源性果糖可能是它驱动肾脏疾病的一个潜在机制[49,79]。
在急性肾损伤患者中,发现了高水平的尿果糖。同样,在缺血性急性肾损伤小鼠的肾皮质中观察到醛糖还原酶、山梨醇和内源性果糖的浓度增加,表明多元醇通路的显著激活。限制多元醇通路在限制缺血性急性肾损伤和促进肾损伤后恢复方面具有关键作用。果糖激酶缺乏阻断了果糖的代谢,增加了ATP的水平,减少了尿酸的产生,抑制了氧化应激的积累,降低了氧化应激,并减少了肾脏炎症,这表明果糖激酶在急性肾脏疾病中的重要作用。当暴露于果糖激酶抑制剂时,野生型小鼠遭受缺血性急性肾损伤后的肾损伤和功能障碍得到了实质性的改善,这表明单独使用果糖激酶抑制剂或与尿酸抑制剂联合使用可能是限制肾脏疾病或促进肾脏恢复的医学上适当的方法[80,81]。果糖摄入似乎提高了血清尿酸水平,并影响尿液pH和尿酸盐代谢,以及草酸和镁刺激尿结石形成,这表明果糖可能是肾结石的重要贡献者,特别是对于伴随低尿酸排泄率的热应激和代谢综合症参与者[82]。
4. 果糖代谢的分子见解
果糖代谢涉及多种遗传参数。醛缩酶B(ALDOB)的突变可以导致遗传性果糖不耐症。遗传性果糖不耐症患者摄入果糖会引发果糖1-磷酸和ATP的耗尽,导致多种疾病,如肝功能衰竭、肝硬化和范可尼综合征。成年遗传性果糖不耐症患者采用限制果糖的饮食可能会造成潜在的心血管风险[83]。敲除醛缩酶B的小鼠表现出与摄入果糖的遗传性果糖不耐症患者相同的症状[84]。此外,摄入低剂量果糖的醛缩酶B缺陷小鼠与脂肪肝进展有关[85]。果糖诱导的醛缩酶B导致血管中甲基乙二醛水平升高。此外,果糖通过刺激碳水化合物反应元件结合蛋白(ChREBP)和阻断FoxO1/3α信号通路,提高了AldoB表达和甲基乙二醛的产生,有助于血管重塑[86]。
酮己糖激酶是果糖代谢中第一个重要的酶,由KHK基因编码,催化果糖转化为果糖-1-磷酸。KHK缺乏负责基本果糖尿,其特点是摄入果糖后血液和尿液中果糖水平升高。基本果糖尿可以在酮己糖激酶敲除小鼠中构建,它改变了果糖代谢的过程,并为预防饮食引起的肝脏脂肪变性和胰岛素抵抗提供了可能[87,88]。KHK-A和KHK-C是两种选择性剪接的KHK蛋白异构体。KHK-A广泛表达,但由于对果糖的亲和力较低,其活性相对较低。相反,KHK-C主要在肝脏、肾脏和肠道中表达,对果糖有更高的亲和力[65,89,90]。果糖诱导的NAFLD导致个体中果糖-1-磷酸的积累,因为果糖激酶将果糖磷酸化的速度比醛缩酶B裂解果糖-1-磷酸的速度快,这表明抑制果糖激酶可能是治疗非酒精性脂肪肝病或遗传性果糖不耐症的潜在治疗方法[91,92]。KHK敲除有助于消除果糖诱导的脂质生成进展,并防止小鼠小肠中果糖诱导的胰岛素抵抗和肝脏脂肪变性[93]。通过抑制或缺失KHK-C而不是KHK-A来保护遗传性果糖不耐症患者免受肠道和肝脏损伤以及低血糖是令人满意的[56]。
碳水化合物反应元件结合蛋白(ChREBP),一种转录因子,在肝脏和小肠中的脂肪生成和糖异生中扮演着关键角色。肠道ChREBP敲除由于与果糖代谢和运输相关的基因表达诱导不足,导致果糖不耐受。过量摄入果糖有助于在ChREBP敲除小鼠中发展以腹泻为主的肠易激综合征。用极端果糖饮食喂养ChREBP敲除小鼠可以构建一个因果糖吸收不良的适当肠易激综合征动物模型[70]。果糖摄入有助于肝脏ChREBP的激活,通过上调BDK和下调PPM1K来磷酸化并激活ATP柠檬酸裂解酶,最终导致脂肪酸的积累和血脂异常[94]。ChREBP作为葡萄糖代谢的关键传感器,在脂肪组织和肝脏组织的脂肪生成以及能量和葡萄糖代谢的调节中起着至关重要的作用[95]。在1型糖原贮积病中,由于葡萄糖-6-磷酸酶(G6PC)的原因,ChREBP的活性增加。肝脏ChREBP的敲低通过抑制VLDL-TG的脂化有力抑制了VLDL-TG的分泌。为了限制非酒精性脂肪肝病的发展,肝脏ChREBP通过调节脂肪酸氧化、脂肪生成和VLDL-TG分泌,保持1型糖原贮积病肝脏中的糖原和甘油三酯平衡[96]。
5. 果糖输注对高尿酸血症的影响
果糖输注与尿酸代谢有着显著的相关性,尿酸的增强积累与高尿酸血症的发展有关。果糖给药导致血清尿酸水平升高和尿液中尿酸排泄增加。在年轻男性中,果糖输注对血清尿酸没有明显影响。有趣的是,在非痛风患者中,果糖只能引起血清尿酸的适度增加。相比之下,在痛风患者中,果糖引起的血清尿酸变化更为明显且持久[97]。
果糖摄入可以引发一系列生化反应,如高弹性酸血症、血清无机磷酸盐降低和血糖降低,这主要是因为磷酸化的果糖转化为果糖-1-P,将进一步参与糖酵解途径[98]。果糖摄入可以促进ATP降解为AMP,导致尿酸的产生并改善血液中的尿酸水平。此外,嘌呤从头合成有助于进一步加速尿酸的产生,这是嘌呤降解的最终产物[99]。更具体地说,果糖激酶介导果糖代谢中果糖到果糖1-磷酸的磷酸化。果糖激酶C的高表达似乎会降低ATP、尿酸产生和核苷酸修饰,有助于肝脏脂肪[13,27]。值得注意的是,该反应过程非常迅速,以至于磷酸盐和ATP的水平大大降低。此外,醛缩酶B介导果糖1-磷酸分解为二羟丙酮磷酸和D-甘油醛的反应速率较慢。每当摄入高水平的果糖时,果糖1-磷酸的积累和细胞内磷酸盐的减少就变得更加容易。而且,细胞内磷酸盐的减少可以激活AMP脱氨酶介导的AMP向肌苷一磷酸的降解,增强嘌呤的降解速率。
简而言之,果糖代谢有效地促进了尿酸的产生。它减弱了尿酸的排泄,而尿酸的过度产生和排泄不足是痛风和高尿酸血症的重要贡献因素[49]。果糖代谢主要发生在肝脏,ATP耗竭广泛涉及各种肝脏代谢过程。ATP耗竭导致细胞内磷酸盐的耗竭。它增强了AMP的水平,这将进一步加强AMP脱氨酶介导的AMP降解为尿酸的过程,并刺激嘌呤核苷酸分解产生的尿酸的产生[47]。此外,由果糖引起的胰岛素抵抗和高胰岛素血症可能通过减少尿酸排泄来增加尿酸水平[16,31,100]。果糖诱发的高尿酸血症与肾脏尿酸排泄减少有关[101]。尽管并非所有研究都指出果糖摄入增加了痛风或高尿酸血症的风险,但我们不能忽视高果糖摄入可能导致的健康后果[22,[28], [29], [30]。
6. 结论
在过去几十年中,由于尿酸浓度升高,高尿酸血症的发病率呈现稳定上升趋势。大量证据表明,高尿酸血症这种代谢疾病显著增加了痛风的可能性。它经常与各种疾病一起发生,包括代谢综合症、高血压、心肌梗死和心力衰竭。果糖摄入是高尿酸血症的一个风险因素,但似乎还没有足够的证据支持减少果糖摄入作为治疗高尿酸血症的方法。需要进一步的实验和临床研究来阐明持续暴露于果糖与高尿酸血症发病率和流行之间的关系。深入了解果糖的摄入、吸收和代谢可能会提供关于果糖摄入对高尿酸血症发病机制影响的基本见解。通过加强对果糖可能导致疾病的潜在机制的认识,将为高尿酸血症提供全面的治疗方法。
https://blog.sciencenet.cn/blog-41174-1440266.html
上一篇:衰老神经元驱动的大脑衰老
下一篇:氯胺酮缓释片抗抑郁作用更好
