博文
哺乳动物去乙酰化酶sirtuin的生物学特征及疾病相关性
||
哺乳动物去乙酰化酶sirtuin的生物学特征及疾病相关性
Mammalian sirtuins: biological insights and diseaserelevance
哈佛医学院病理系格伦衰老分子生物学实验室 Marcia C. Haigis David A. Sinclair
摘要:衰老过程伴随健康机体多个器官系统的功能性下降,并导致疾病(如2型糖尿病、神经变性疾病、癌症和心血管疾病)发病率和死亡率增加。长期以来,研究者聚焦于孤立器官的自身通路研究,将其作为发现疾病根本病因的策略,希望由此设计出更好的药物。酵母衰老研究促成了保守性酶类家族sirtuin的发现。sirtuin可影响延长寿命的多条通路,并增强机体的健康水平。自从10年前第一个哺乳动物sirtuin(即SIRT1)发现以来,我们对sirtuin酶学特征的认识取得了重要进展,包括其调节作用,以及改善哺乳动物生理功能和健康水平的作用。本文将总结并讨论过去十年来的研究进展,以及该领域未来面临的挑战。
关键词:染色质(chromatin);代谢(metabolism);去乙酰化酶(deacetylase);肿瘤(cancer);心血管疾病(cardiovascular);炎症(inflammation)
Haigis MC, Sinclair DA. Mammaliansirtuins: biological insights and disease relevance. Annu Rev Pathol 2010, 5:253–295
整整10年前,研究者发现沉默信息调节因子2(Silent Information Regulator 2,SIR2)基因可通过抑制基因组不稳定性,延长芽殖酵母的寿命[1,2]。此后发现,SIR2样基因(称之为sirtuin)可见于大多数生物体(包括植物、细菌和动物),对于促进机体健康和存活发挥关键作用[3]。其中的重要发现包括,sirtuin如何从分子水平感知能量的摄入量、日照时间的变化,以及环境的应激;如何通过诱导相关变化来作出应答,以促进逆境条件下的存活。研究还发现,sirtuin可进行烟酰胺腺嘌呤二核苷酸(NAD+)依赖性去乙酰化反应[4]。由此开启了一系列有关sirtuin调节代谢的研究,其中涉及小分子对sirtuin活性的调节作用。针对从酵母到哺乳动物的各类物种,通过限制热量或药物干预,均可激活sirtuin以延长寿命和促进健康,这表明以sirtuin为靶点可望开发出治疗增龄性疾病的药物[5]。本文将综述过去10年来sirtuin从分子、细胞和机体水平,影响哺乳动物生理功能的相关研究;还将讨论本领域未来面临的挑战。
sirtuin生物学
衰老调节因子SIR2/SIRT1的发现
Sir2蛋白来源于酿酒酵母(Saccharomyces cerevisiae,又称芽殖酵母),是NAD+依赖性去乙酰化酶和ADP-核糖转移酶家族(称之为sirtuin家族)的早期成员[6]。最初发现,Sir2酶编码基因SIR2对酵母的交配型基因座(mating-type loci)具有转录沉默(transcriptional silencing)效应。随后发现,SIR2针对酵母的端粒和重组DNA(rDNA)的沉默也发挥关键作用[7,8],对羰基化蛋白在母细胞与子细胞之间的分离也具有重要的沉默效应[9]。
sirtuin在衰老中的作用,最初通过酵母的复制寿命(replicative life span)模型而发现,该模型可测量酵母母细胞在衰老前产生子细胞的次数。1997年发现,酵母衰老的原因源自rDNA基因座的重组。经同源重组,染色体外rDNA环(extrachromosomalrDNA circle,ERC)形成是引发衰老母细胞ERC扩增的起始事件,而母细胞ERC复制和优先分离(preferential segregation)则是导致其衰老的因素[1]。1999年发现,额外增加SIR2基因拷贝,可抑制rDNA重组、减少ERC形成,从而延长复制寿命约30%;而删除SIR2则ERC形成增多,复制寿命缩短[2]。
|
图1 sirtuin去乙酰化反应及应激和营养对其调节作用 |
与Ⅰ型和Ⅱ型去乙酰化酶(水解底物的乙酰基)不同,sirtuin去乙酰化酶的催化反应需经过两个步骤,即消耗烟酰胺腺嘌呤二核苷酸(NAD+),释放出烟酰胺(NAM)、O-乙酰基-ADP-核糖(AADPR)和去乙酰化底物。酰胺基到酯的转酰基作用并无益处,而NAD+水解则可对推进整体sirtuin反应发挥有益功效。有证据支持以下机制,即ADP-核糖转移反应中通过亲电子作用捕获乙酰基中的氧,从而形成ADP-核糖-肽酰亚胺复合体(ADP-ribose peptide-imidate complex)。这一中间体可能仅存在数秒钟,却足以使NAM进入C形口袋,从而催化一个逆反应。通过清除NAM,或将其转化为NAD+可促进激活sirtuin的过程;而NAM转化为NAD+需要PNC1[烟酸磷酸核糖转移酶,存在于酵母和简单后生动物(metazoan)]或NAMPT[NAM磷酸核糖转移酶,存在于哺乳动物和α-变形菌(proteobacteria)],两种基因可因应激和营养限制而上调。 |
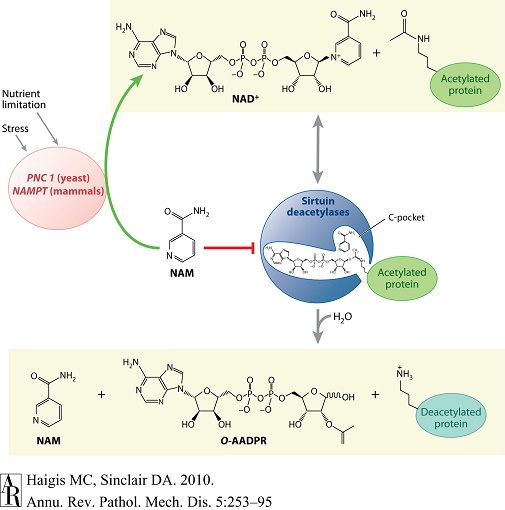
1990年代末期以来,对酵母SIR2基因的研究已渐趋明朗。已知该基因属于一个保守性基因大家族,可见于从细菌到哺乳动物的众多物种[10]。通过对多种较复杂模式生物(如秀丽隐杆线虫Caenorhabditis elegans和果蝇Drosophila)的研究,已明确SIR2在衰老过程中发挥保守性作用。在线虫,sir-2.1基因延长寿命需要叉头蛋白(forkhead protein)DAF-16的参与,但并不需要完整的胰岛素信号通路[11,12]。在应激条件下,sir2.1直接与DAF-16结合,并将后者激活[12]。并且,sir-2.1似乎并不对胰岛素信号的变化作出反应;sir-2.1只是在经应激处理(如热刺激和氧化损伤)时才被激活。而增加果蝇的SIR2直系同源基因(dSIR2)的拷贝数量,也可延长寿命[13]。有趣的是,仅在果蝇神经元中特异性过表达dSIR2,即足以延长其寿命。
哺乳动物sirtuin的酶活性及其调节
通过增加SIR2基因拷贝数量可延长酵母、线虫和果蝇的寿命,这些激动人心的发现激发了人们对哺乳动物sirtuin的研究。sirtuin是否可延长哺乳动物的寿命?哺乳动物sirtuin是否可促进健康并保护机体对抗增龄性疾病?这些蛋白是否在热量限制(CR)过程中发挥重要的介导作用?sirtuin的靶点有哪些?上述核心问题推动着这一领域的研究。仅经过10年,哺乳动物sirtuin研究即取得了可喜的进展。然而,我们对哺乳动物sirtuin与衰老和增龄性疾病相关性的研究才刚刚开始,对于众多sirtuin的理解还处于初级阶段。
哺乳动物sirtuin存在高度保守的NAD+结合和催化结构域(称为sirtuin核心结构域)[14],由此可将sirtuin分为7类,即SIRT1-7(表1)。虽然sirtuin具有相对保守性,但由于其氨基端和羧基端仍存在差异,故其可能具有高度分散的生物学功能,具体原因为:(1)具有不同的酶活性;(2)可与伙伴蛋白和底物特异性结合;(3)具有独特的亚细胞定位和表达模式[15]。
表1 哺乳动物sirtuin概况 | ||||
sirtuin | 定位 | 相互作用 | 生物学功能 | 基因缺失表型 |
SIRT1 | 细胞核 | FOXO、PGC-1α、NF-κB、Ku70等 | 代谢、应激 | 发育缺陷,某些遗传背景下为致命性 |
SIRT2 | 胞浆 | 微管蛋白、H4、FOXO | 细胞周期 | 发育正常 |
SIRT3 | 线粒体 | AceCS2、GDH、复合体Ⅰ | 生热作用(Thermogenesis)、ATP产生 | 发育正常 |
SIRT4 | 线粒体 | GDH、IDE、ANT | 胰岛素分泌 | 发育正常 |
SIRT5 | 线粒体 | CPS1 | 尿素循环 | 发育正常 |
SIRT6 | 细胞核 | 组蛋白H3、NF-κB | 碱基切除修复 | 早衰 |
SIRT7 | 核仁 | Pol Ⅰ | rDNA转录 | 体型较小,短寿命,心脏缺陷 |
缩写 AceCS2:乙酰CoA合成酶2;ANT:腺苷酸转运体;CPS1:氨甲酰磷酸合成酶1;FOXO:叉头框蛋白O;GDH:谷氨酸脱氢酶;IDE:胰岛素降解酶;NF-κB:核因子κB;PGC-1α:过氧化物酶体增殖物激活受体γ(PPARγ)辅激活因子1α;Pol Ⅰ:聚合酶Ⅰ;rRNA:重组RNA | ||||
首先发现的sirtuin标志性反应为核糖转移反应(ribosyltransfer reaction)。研究显示,经细菌sir2同源产物cobB的催化,5,6-二甲基苯并咪唑(5,6-dimethylbenzimidazole)可被转化为α-核糖-5,6-苯并咪唑(α-ribose-5,6-benzimidazole)[16],由此引发了一项令人惊奇的发现,即sirtuin为NAD+依赖性去乙酰化酶和ADP-核糖转移酶。已知多数sirtuin均具有NAD+依赖性去乙酰化作用(图1)[4,17-19],而SIRT4具有NAD+依赖性的单ADP-核糖转移酶活性[20,21],SIRT1和SIRT6则显示兼有自主的ADP-核糖转移酶活性和底物特异性去乙酰化活性[22]。SIRT4和SIRT7研究尚未观察到去乙酰化酶活性,但此活性可能需特异性底物(与SIRT6类似)才能显示。虽然酵母SIR2为组蛋白去乙酰化酶(HDAC),而对其他生物sirtuin的研究却发现了一大批非组蛋白底物。sirtuin针对乙酰基修饰的赖氨酸残基发挥去乙酰化作用,且在各反应周期,均需借助NAD+剪切这一独特的酶学机制方可实现[4,18,23-25]。因此,不同于水解乙酰赖氨酸残基的其他HDAC,sirtuin活性与细胞的代谢状态密切相关。
sirtuin具有精巧和新颖的去乙酰化机制。去乙酰化反应始于对NAD+的酰胺基剪切,从而形成烟酰胺(NAM)与ADP-核糖(ADPR)共价的肽酰亚胺中间体(peptide-imidate intermediate)(图1)。该中间体分解为O-乙酰基-ADP-核糖(AADPR),并释放出去乙酰化的底物[25-27]。酰胺基到AADPR的转酰基作用并无益处,而NAD+水解则可对推进整体sirtuin反应发挥有益功效[28-30]。虽然某些细节有待解释,但已知肽键结合可使酶结构易于发生构象变化,使NAD+与酶的亲核体(nucleophile)发生反应,生成酶稳定的ADPR中间体[30]。
NAD+生物合成对sirtuin的调节
越来越多的证据显示,存在于各种亚细胞器的NAD+生物合成途径,对于sirtuin的调节也具有重要作用。Sternglanz和Sinclair实验室均报道,NAM可抑制sirtuin,且后者以非竞争性方式作用于NAD+(31-33)。非竞争性抑制效应这一奇特发现,促使研究者推测NAM可能结合于sirtuin的一个新部位,且存在生理相关性;随后这一想法得到证实:抑制效应的实现需NAM进入sirtuin高度保守的C形口袋结构(临近NAD+结合位点)[31,34],参与生成肽酰亚胺中间体的反应,从而实现NAD+的再生[33]。这种可逆性的调节反应在生物体内罕有发生,被认为是调节诸多sirtuin的重要机制。
在酵母、线虫和果蝇中,NAM经过四个步骤再生为NAD+,其中第一步经烟酸磷酸核糖转移酶-1(nicotinate phosphoribosyltransferase,Pnc1)催化产生烟酸。在酿酒酵母和黑腹果蝇(D. melanogaster)中,PNC1受到环境应激(如热刺激和CR)时将被上调,以增强对环境应激的抵抗力并延长寿命(35-37)。对此有观点认为,应激和饮食限制促进存活和延长寿命是一种古老的存活反应。另一种NAD+前体——烟酰胺核苷(nicotinamide riboside,NR)可见于酵母和哺乳动物细胞,外源性提供NR亦可延长酵母寿命[38,39]。哺乳动物将NAM再生为NAD+需要两个步骤。第一步,NAM磷酸核糖转移酶[Nampt,或称内脏脂肪素(Visfatin)或前B细胞集落增强因子(PBEF)]将NAM转化为烟酰胺单核苷酸(NMN)[10,41]。第二步,分别位于细胞核、高尔基体和线粒体的同工酶——NMN腺苷酰转移酶Nmnat1、Nmnat2、Nmnat3将NMN再生为NAD+[42]。
与酵母PNC1调节Sir2相一致,哺乳动物Nampt也是SIRT1活性的主要调节因子(40,43-45)。有趣的是,位于Nampt下游的Nmnat1可在启动区与SIRT1直接作用,提示Nmnat1可将NAD+转移至SIRT1,或存在纳米级NAD+池以影响SIRT1活性[45]。
Nampt酶和NMN也可见于小鼠和人类血浆(此时该酶称为eNAMPT)[46]。Imai和同事[47]提出,NMN为一信号分子,遭受应激和营养缺乏的细胞,可借助其与机体其他部位实现交流。这一观点可称为“NAD的世界”(NADworld),研究者对这一领域有相当浓厚的兴趣,特别是有可能采用NMN或其下游分子(如NR)作为治疗2型糖尿病(T2DM)或其他增龄性疾病的手段。
SIRT1和Nampt是哺乳动物昼夜节律(circadian clock,生物钟)反馈循环的主要成分。Nampt受CLOCK-BMAL-SIRT1复合体的转录调节,而后者可加强NAM到NAD+的转换;进而则激活SIRT1,后者再重新激活Nampt的表达——以上均在12小时循环周期内完成[50-53]。由于NAD+水平受到动态调节,因此获取和比较其在一日内不同时点的结果时,应当谨慎。
在哺乳动物,NAD+不仅可被sirtuin和多聚ADP核糖聚合酶(PARP)破坏,也持续受到CD38(一种甘油水解酶)的分解。最初将CD38描述为一种在细胞表面与免疫相关的NAD环化酶,但只具有较轻微的活性。基因抑制或删除CD38可增加稳定态NAD+的水平,故有人推测抑制此酶是激活sirtuin的有效手段[54,55]。如下所述,缺乏CD38的1岁小鼠可免受增龄性肥胖和糖尿病的侵袭,或许即是由于sirtuin活性的增强[54,55]。
sirtuin反应产生的AADPR是一种新型代谢产物,但对其研究较少,可能具有重要的生理作用。例如,酵母的AADPR可促进Sir复合体(特别是Sir3与Sir2/Sir4二聚体)的组装,并促进Sir复合体扩散到染色质[56]。而在哺乳动物,AADPR可结合并激活瞬时感受器电位M型(transientreceptor potential melastatin,TRPM)相关通道2[57]。AADPR在体外可被nudix水解酶所代谢,而在体内则可能被一种未知酶代谢为乙酸[58]。因此,在sirtuin介导的去乙酰化过程中,NAD+剪切产物所生成的代谢物可能介导了与衰老和代谢有关的生物学功能。尽管sirtuin生物学领域对这一领域所知尚少,但未来数年中则可能有某些更有趣的发现。
哺乳动物sirtuin的亚细胞定位
哺乳动物sirtuin可见于多个细胞区室。SIRT1、SIRT6和SIRT7主要见于细胞核[59],SIRT3-5位于线粒体,而SIRT2主要见于细胞浆。这些蛋白的亚细胞定位可能与细胞类型、应激状态和分子间相互作用有关。例如,SIRT1和SIRT2均可见于细胞核和细胞浆,且与同样的核蛋白和胞浆蛋白相互作用[60-62]。
何种原因控制着sirtuin的定位?为何SIRT1有时位于核内,有时位于胞浆?原因首先是各种sirtuin具有的主要氨基酸信号序列促成了其亚细胞定位。例如,SIRT1、SIRT6和SIRT7定位于细胞核,很大程度上是由于其具有核定位信号。SIRT1除含有两个核定位信号区外,还有两个核输出信号[61]。因此,核定位信号与核输出信号即可支配SIRT1的胞浆定位和核定位。
SIRT3–5含有氨基端线粒体靶向序列,故一般认为其定位于线粒体基质[21,22,63-65];不过并未排除这些蛋白定位于线粒体的其他区室。关于SIRT3的定位就曾有激烈的争论[66]。最初报告发现SIRT3只定位于线粒体[22,64],而随后研究提示,细胞应激时SIRT3可能会从线粒体转位至核内[67,68]。所以深入理解SIRT3的结构和酶学特性,才有可能揭示其功能。最近一项研究通过一种硫代乙酰肽(thioacetylpeptide)的诱捕(trap),鉴定了SIRT3脱辅酶(apoenzyme)和反应中间体(reaction-intermediate)的晶体结构[69]。该研究显示,底物可诱导SIRT3的构象变化,而该乙酰化肽即先于NAD+与SIRT3结合,成为其首个底物[69]。总之,sirtuin可分布于多个细胞区室,其定位可能因组织/细胞类型和生理状态而呈动态化。
热量限制过程中sirtuin的作用
多项研究显示,低热量饮食(即CR)可使多个物种的寿命延长50%以上,包括酵母、线虫、果蝇和小鼠[70]。然而也有一些例外,比如野生小鼠和家蝇[71,72]。而对于哺乳动物,CR也可引发葡萄糖稳态改善这一生理变化,具体为通过对啮齿动物和人类进行CR,显示其胰岛素和葡萄糖水平降低,而胰岛素敏感性改善。这些代谢变化与衰老相关,因为模式生物研究显示,寿命的调节涉及到胰岛素信号的降低[73]。关于CR的生理学研究已有详细综述[70]。
现已发现Sir2是酵母、线虫和果蝇寿命的保守性调节因子,同时已知sirtuin活性依赖于NAD+,由此加深了人们对Sir2在CR中作用的研究兴趣。是否Sir2活性介导了CR的有益效应?答案取决于CR方案(适度限制还是强烈限制)和遗传背景。多项酵母复制衰老(母细胞分裂次数)研究显示,CR延长寿命确实需要SIR2参与。并且,适度的CR饮食(0.5%葡萄糖)可增强线粒体呼吸,上调Sir2活性,并抑制rDNA重组[36,74]。而酵母Sir2的同源蛋白Hst1和Hst2,也显示可延长其复制寿命[75]。
然而,对于sirtuin在酵母CR中的作用仍有部分争论。更强烈的CR方案(0.05%葡萄糖)也可延长酵母复制寿命,但并不需要SIR2或线粒体呼吸的参与[76,77]。Lamming和同事研究显示,同时删除SIR2和HST2可阻断CR介导的寿命延长,提示酵母sirtuin家族存在功能冗余(functional redundancy)。在饥饿条件下,SIR2对于酵母存活(时序寿命,chronological life span)并无作用,对某些极长寿命的突变品系(如sch9)却显示可降低其存活[78]。而在线虫CR模型中,sir-2.1似乎并非延长寿命所必须;当然如果试验其他的CR方式,sir-2.1也可能会与某种饮食相关,线虫另外的三种sirtuin也可能会有涉及。
因为酵母Sir2蛋白数量在CR期间并不增加[36],研究人员开始寻求Sir2活性增强的其他解释。Guarente实验室[80]认为,CR期间还原型烟酰胺腺嘌呤二核苷酸(NADH,为SIR2抑制剂)减少会导致Sir2活性增强,而Sinclair和Smith实验室则提出Sir2活性增强源自CR期间PNC1上调,这期间将耗竭NAM并经NAD+补救途径(salvage pathway)增加其流出量。有趣的是,延长寿命的温和应激,如升高温度(37℃)或氮气限制,均可使PNC1上调。这些研究资料似可提示,CR具有毒物兴奋效应(hormesis),或属于一种温和应激,从而诱导有益的防御反应(83-85)。这其中NADH与PNC1的机制有根本的不同:前者直接调节sirtuin,而后者与应答特定应激的活跃遗传学通路有关。2003年以来,有关酵母和哺乳动物中Sir2/SIRT1调节机制的研究证据日渐增多,提示NADH与PNC1可能有协同作用[32,86,87],而某些研究者则质疑NADH在体内是否足以强效抑制SIRT1[26]。
上述研究大大激发了对哺乳动物CR期间sirtuin作用的研究兴趣。目前,多数研究均涉及到SIRT1,且研究模型证实SIRT1介导CR的多种有益效应。最初有啮齿类动物研究显示,CR上调多种组织中SIRT1的表达,如脑、肾、肝脏、白色脂肪和骨骼肌[88]。然而,SIRT1被诱导的现象并非见于所有CR研究,其可能仅以组织特异性方式被诱导,甚至会降低[89]。值得注意的是,某些组织的NAD+水平在CR期间也会增加。因此,CR期间多种组织可并见SIRT1蛋白水平和NAD+浓度升高(或经NAD补救途径流出),共同促进SIRT1活性增加。
小鼠研究进一步强化了SIRT1与CR之间的关联性。Guarente实验室[90]首先报道,CR诱导某种表型(身体活动量增加)需要SIRT1参与。过表达SIRT1的转基因小鼠也显示多种代谢益处,且与CR表型重叠。例如,经β-肌动蛋白(β-actin)启动子驱动,建立表达SIRT1的基因敲入小鼠模型;与野生型对照组比较,显示过表达SIRT1的小鼠瘦小,糖耐量增加,且血胆固醇、脂肪因子(adipokine)和胰岛素水平降低[91]。另一项精巧的系列研究显示,采用自身启动子的野生型或SIRT1突变小鼠,均可通过细菌人工染色体(bacterialartificial chromosome,BAC)过表达SIRT1。此类小鼠模型显示,升高的SIRT1并未改善基础糖耐量,而是缓解了肥胖诱导的糖耐量异常[92]。另一项研究显示,SIRT1转基因小鼠可耐受肝脏脂肪变性(脂肪肝)和胰岛素抵抗[93]。这些小鼠模型的SIRT1表型,与给予SIRT1激活剂[如白藜芦醇(resveratrol)和SRT1720]小鼠的反应相类似[94-96]。由于这些研究多数采用全身性转基因动物,如能发现导致上述保护表型的特定组织将会大有裨益。另一个有趣的问题是,过表达SIRT1是延长平均寿命、最大寿命,还是仅健康寿命(health span)?
SIRT1缺乏小鼠研究也证实,SIRT1介导了CR部分表现。如上所述,前一项研究中观察到喂饲CR饮食小鼠的行为变化[90],即CR导致野生型小鼠活动量增加。处于野生环境且食物匮乏时,这种效应可能会促进动物觅食,从而有助于其存活。全身SIRT1缺乏小鼠并未见其活动量增加,提示上述CR表型需要SIRT1参与[90,97]。近期已测定了SIRT1缺乏小鼠的寿命,显示这种小鼠的寿命短于同窝出生(littermate)的野生型小鼠,CR并不会延长这种小鼠的寿命[98]。上述研究结果提供了重要证据,证实在哺乳动物中SIRT1可能为CR延长寿命必需物质。然而要作出肯定的结论却还有更复杂的影响因素,因为有发现存活至成年的SIRT1缺乏小鼠会出现早死,还伴有发育缺陷[99]。若进一步采用组织特异性SIRT1敲除小鼠,可能会确定何种组织造成了此种重要表型。另外值得注意的是,很少有研究探索SIRT2-7在CR表型中的作用,但这些蛋白在调节CR生理反应过程中可能也会发挥一定作用。
小分子、蛋白相互作用和修饰作用对sirtuin的转译后调节
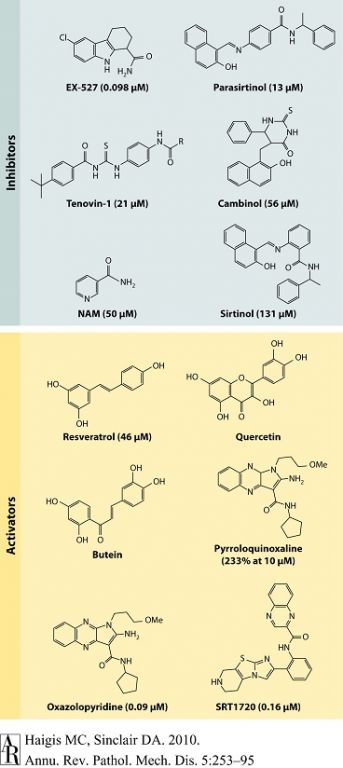 |
图2 SIRT1化学抑制剂和激活剂 |
过去10年来,报道了多种类型的SIRT1激活复合物(STAC)或抑制剂。括号内为该物质针对SIRT1已知的半抑制浓度(IC50)和1.5倍激活浓度(EC1.5)。所有抑制剂中,只有烟酰胺(NAM)为生理性抑制剂,而NAM的类似物虽然也可激活sirtuin,但明显是通过阻断C形口袋而实现(见图1)。多酚类激活剂(如白藜芦醇)似乎也是通过相同机制结合于相同位点以激活sirtuin(即主要通过降低米氏常数而起效),更强效的激活剂(如SRT1720)亦是如此。抑制作用的效价以IC50表示(抑制50%活性的浓度),激活作用效价以EC1.5表示(激活1.5倍活性的浓度)。 |
考虑到SIRT1水平可对环境刺激(如日光、细胞应激和CR)作出反应,故容易理解其基因表达受到多种转录因子和microRNA(包括p53、FOXO3、HIC1:CtBP、E2F1、c-Myc、miR-34和miR-199)的调节[100-109]。而对其他sirtuin在转译水平如何受到调节,尚所知甚少。
如上所述,sirtuin还受到转译后调节。已发现sirtuin可受到小分子直接调节或经蛋白相互作用调节,这便有可能模拟CR的有益功效,而不必限制热量。小分子的SIRT1调节剂有splitomycin、sirtinol和EX-527[110],以及抑制酵母SIR2、SIRT1和SIRT2的sirtinol类似物(图2)[111]。SIRT1激活复合物(STAC)包括白藜芦醇、SRT1720和SRT2183,以及NAM类似物[94,112,113]。除NAM类似物(如甲基NAM)干扰NAM对sirtuin的抑制作用外[32],其他SIRT1激活剂的作用原理是降低底物和NAD+的米氏常数(Michaelisconstant,Km),并小幅增加酶反应速度(Vmax)[94,113]。
10年来已合成和分析了3500多种STAC[114],其中白藜芦醇及结构类似的其他天然多酚类,如漆黄素(Fisetin)、紫铆花素(butein)(图2)均显示可延长酵母、果蝇和肥胖小鼠等多个物种的寿命[115-118];但也有部分实验室报告其(几乎)没有延长寿命作用[119,120]。问题依然是,这些分子如何激活Sir2和SIRT1?通过荧光法或质谱分析法等检测手段试图探测其激活效应,并采用典型的肽类底物,这些底物含有额外的基团如7-氨基-4-甲基香豆素(7-amino-4-methylcoumarin,AMC)、四甲基罗丹明(tetramethyl-rhodamine,TAMRA)[94,113,121];然而却发现这些基团对于探测小分子对SIRT1的激活效应并非必要[122]。某些研究还怀疑这些检测手段是否能涵盖激活的SIRT1对天然底物的效应,而越来越多的研究显示这些底物与SIRT1之间存在依赖性,其在体内具有模拟CR和SIRT1过表达的能力[95,112,116,123,124];故而更多研究者认为这些底物在很大程度上通过SIRT1起作用[92,93,96,125]。Sauve[28]推测检测中采用的荧光团(fluorophore)可再现细胞天然底物的生物物理学特征,从而提供了活体内与白藜芦醇等化合物结合的同源位点。
目前已发现多种SIRT1调节剂,其中激动剂有SIRT1主动调节剂(active regulator of SIRT1,AROS)[126]、necdin[127],抑制剂有乳腺癌缺失蛋白1(deleted in breast cancer 1,DBC1)(图3)[128,129]。其中DBC1和AROS均可结合于SIRT1的氨基端,该位点也是SIRT1的小分子激活因子的结合位点,故而两者间可能有共同的调节机制。现已明确sirtuin还可被直接修饰。例如,SIRT1在体内至少有13个磷酸化的氨基酸残基位点,而除去这些磷酸盐后将降低其催化活性。若细胞周期蛋白B/细胞周期蛋白依赖性激酶1(cyclinB/CDK1)的2个结合位点(苏氨酸530和丝氨酸540)发生突变,将会破坏正常的细胞周期和增殖[130]。上述SIRT1活性调节剂在特定疾病模型中的功效将在下文详述。
sirtuin研究的新兴领域涉及代谢调节。维持能量稳态需要能量摄入、储存和消耗的协同调节。在健康个体,这一过程中某一部分的波动,可由另两部分的适应性调节(adaptation)而代偿。这种精巧的平衡通过内分泌途径和代谢途径来协调,涉及到器官间通讯。因此,代谢途径的功能即为感知营养摄入和环境刺激,并作出恰当反应。当对能量摄入、储存和消耗变化的适应能力丧失时,就会发生代谢紊乱。代谢稳态异常会导致严重后果,常见肥胖和T2DM等病症;并且代谢紊乱的某些表现(如胰岛素抵抗)随增龄而频发。阐释调节代谢稳态的核心通路,是sirtuin研究的纵深领域;此项研究对于揭示调节适应性反应的分子网络,以及治疗代谢性疾病,具有重要意义。关于SIRT1和线粒体sirtuin如何调节针对营养物的适应性反应,本部分将综述我们对该问题的理解。
|
图3 增龄性生理变化中SIRT1的调节 |
SIRT1活性的调节可通过烟酰胺腺嘌呤二核苷酸(NAD+)和烟酰胺(NAM)浓度、SIRT1自身水平及其磷酸化而实现;SIRT1可被SIRT1主动调节剂(AROS)激活,被乳腺癌缺失蛋白1(DBC1)抑制。激活SIRT1可促进神经元存活,防止心肌细胞死亡。营养剥夺期间,肝脏SIRT1可通过LXR、PGC-1α和PPARα促进脂肪酸氧化和糖异生。在白色脂肪组织(WAT),SIRT1可通过抑制PPARγ而降低脂肪储存。SIRT1还分别可通过抑制UCP2和与FOXO相互作用,以促进胰岛素分泌和胰腺β细胞存活。骨骼肌SIRT1可通过激活PGC-1α促进线粒体的生物合成。改引自参考文献[15]。 缩写 CNS:中枢神经系统;FOXO:叉头框转录因子O亚组;LXR:肝X受体;NF-κB:核因子-κB;PGC-1α:过氧化物酶体增殖物激活受体γ(PPARγ)辅激活因子1α;PPARα:过氧化物酶体增殖物激活受体α;UCP2:解偶联蛋白2。 |

针对sirtuin在代谢途径中的作用,已进行了深入的研究。同样,多数研究均聚焦于SIRT1。虽然对于SIRT1的作用范围和细节尚未完全阐明,但有充分证据提示该酶可感知营养的可获得性,并将此信息传递给负责能源物质利用和能量适应性的蛋白。SIRT1的多个靶点及其调节机制如图3所示。SIRT1可与某些重要的转录因子结合,并使其去乙酰化,这些转录因子包括过氧化物酶体增殖物激活受体γ(PPARγ)、PPARα、PPARγ辅激活因子1α(PGC-1α)、叉头框蛋白O(FOXO)转录因子家族;进而会促进代谢反应,如胰岛素分泌、糖异生(gluconeogenesis)和脂肪酸氧化。所以SIRT1是一个具有多效性的能量感受器,可辅助机体进行适当的生理反应。
线粒体生物合成(biogenesis)与糖异生
这方面的生化和细胞研究数据,可通过模型小鼠的体内研究获得支持。例如,PGC-1α可能为SIRT1的重要靶点,并见于多种细胞类型。而最初发现并克隆PGC-1α时,是将其作为棕色脂肪组织中由寒冷诱导的PPARγ激活因子[131]。现已明确,在多种细胞类型中,PGC-1α是为产生能量而进行线粒体生物合成及底物摄取和利用的强效诱导物[132]。一项精巧的系列研究首先描述了SIRT1–PGC-1α网络在肝细胞中重要作用[132]。在体外,SIRT1可与PGC-1α相互作用并使PGC-1去乙酰化[132],由此导致后者活性上调。
激活的SIRT1不仅使PGC-1去乙酰化,还可致SIRT1依赖性诱导糖异生基因,并抑制糖酵解基因[132]。该研究采用腺病毒特异性降低成年小鼠肝细胞SIRT1水平,由此SIRT1与糖异生的相关性得以证实[132]。此模型通过基因抑制肝SIRT1,导致轻度低血糖和葡萄糖生成增加。而对这些成年动物来说,肝SIRT1水平降低还可改善葡萄糖清除率并增强胰岛素敏感性。与之类似,基因抑制成年大鼠肝SIRT1,可减少T2DM大鼠模型的葡萄糖生成[133]。这些研究中,通过禁食可见小鼠肝NAD+浓度和SIRT1水平升高[132]。多家研究小组也报告,营养剥夺(如禁食或CR)期间,肝NAD+升高(测自全细胞抽提物或线粒体)[44]。
而另外两项研究采用肝特异性SIRT1缺乏小鼠发现,胰岛素稳态、葡萄糖稳态,以及糖异生均无变化[89,134]。造成这些模型差异的原因何在?可能是因为绝对缺乏SIRT1的模型会激活代偿途径,从而使糖异生恢复;也就是说,这些小鼠看似发育正常,但在肝脏发育过程中,因缺乏SIRT1激活了代偿机制。另外一种解释是,肝脏SIRT1无法诱导环磷酸腺苷反应元件结合蛋白(CREB)辅激活因子2(TORC2)提供的正常反馈环,该现象见于近期Montminy和同事的抑制SIRT1刺激性糖异生研究[135]。总的来看,以上研究资料说明,针对哺乳动物的单一组织进行解释性研究,具有相当的难度和复杂性。
随后研究显示,SIRT1主要通过调节PGC-1α活性,来协调线粒体底物利用的变化。这一转变过程使细胞保留葡萄糖而氧化脂肪酸,这可联系到营养剥夺所见的底物利用情况。在骨骼肌,SIRT1使PGC-1α去乙酰化以促进脂肪酸氧化基因的转录;在低营养情况下,要转变为脂肪酸氧化方式正需要SIRT1参与[136-139]。另外,激活PGC-1α还会促进氧化磷酸化基因的转录,提示SIRT1可能与能量产生过程的脂肪酸氧化相关联[140]。与这些发现相一致,全身SIRT1缺失小鼠亦显示,其肝脏线粒体呼吸率和活性氧(ROS)生成量均较低[97]。并且,SIRT1缺失小鼠在24h禁食后,可见AMP激活的蛋白激酶(AMPK)水平升高,提示小鼠的ATP水平降低[97]。因此,激活PGC-1α可能正是SIRT1调节线粒体生物合成的直接机制。
|
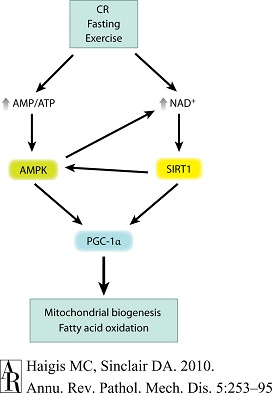
图4 SIRT1-AMPK代谢调节网络 |
感知性能量剥夺(perceived energy deprivation),如禁食、热量限制(CR)和运动,可升高AMP/ATP比值,激活AMP激活蛋白激酶(AMPK)。能量剥夺还可升高烟酰胺腺嘌呤二核苷酸(NAD+)水平,激活NAD+依赖性SIRT1去乙酰化活性。激活的AMPK和SIRT1可分别将过氧化物酶体增殖物激活受体γ(PPARγ)辅激活因子1α(PGC-1α)磷酸化和去乙酰化,从而诱导线粒体生物合成和脂肪酸氧化。上述通路存在交互效应(cross talk),即AMPK激活可升高NAD+,而SIRT1也可激活AMPK。 |
营养剥夺期间,骨骼肌SIRT1被激活可致整体转变为肝脏脂肪酸氧化方式[134,138]。SIRT1可能是通过作用于PGC-1α,以激活PPARα转录,从而诱导脂肪酸分解基因表达[134]。过表达SIRT1转基因小鼠体重增加的情况与野生型类似,但却可抵抗高脂饮食所致的脂肪肝[93],这与SIRT1激活剂白藜芦醇和SRT1720的功效一致[96,138,141,142]。SIRT1缺失肝细胞则显示较低的脂肪酸氧化率[134]。并且,特异性肝SIRT1敲除小鼠喂饲高脂饮食、西式饮食后,其体重较同窝出生对照鼠增加,且空腹血脂水平升高,酮体水平降低[134]。
当然也有一些相反的证据。某项研究显示,类似的特异性肝SIRT1敲除小鼠喂饲高脂饮食后,体重反而减轻[143]。另一项研究则认为,由于SIRT1可激活肝X受体(LXR),而特异性肝SIRT1敲除小鼠因缺乏LXR激活,导致脂肪生成减少,这可能为高脂饮食未致体重增加的机制之一[89]。总之,上述研究提示肝SIRT1活性在高脂饮食时升高,CR时降低。因此,禁食状态下,sirtuin活性(或NAD+)很可能是在某些组织升高而在其他组织则受到抑制。如何将两项实验协调起来呢?同时采用C57BL/6小鼠研究后,对此的最佳解释便是饮食成分的差异造成了某些自相矛盾之处。实际上,后一项研究使用的碳水化合物成分显著高于其他研究。
重要的是,另有研究已将SIRT1与改善葡萄糖稳态和胰岛素稳态,以及能源物质利用联系起来。例如,采用白藜芦醇或更强效的SIRT1激活剂(如SRT1720和SRT2183)体内激活SIRT1,可诱导基因表达发生变化(与激活肝脏和肌肉PGC-1α所见一致),从而促进线粒体呼吸,并增加胰岛素敏感性[94-96]。这种情况下,糖异生缺乏可能是由于TORC2对肝PGC-1α的抑制效应,而该效应在肝特异性基因改变个体并不存在[135]。与这一发现相一致,培养细胞中阻断SIRT1可致胰岛素抵抗[124]。其研究者认为SIRT1可抑制蛋白酪氨酸磷酸酶1B(PTP1B),而后者为胰岛素信号的负调节因子,这便解释了SIRT1作用于外周组织胰岛素信号的潜在机制[124]。就饥饿反应而言,值得注意的是,诱导全面的自噬反应也需要SIRT1的参与[144],然而尚需通过后续的体内研究以评估这一结果与代谢或细胞存活的相关性。
运动可诱导能量剥夺状态,从而激活骨骼肌中类似的代谢通路。除运动之外,还发现AMPK也可通过升高细胞内NAD+以增强SIRT1活性[87]。随后,SIRT1去乙酰化并激活由PGC-1α、FOXO1和FOXO3驱动的转录程序。这便有助于解释为何激活AMPK和SIRT1会出现许多功能重叠(图4)。采用SRT1720激活SIRT1并不会直接激活AMPK,但可增强胰岛素敏感性,改善糖耐量,增加呼吸商,并使肌肉转向氧化途经[138]。因此,在AMPK未被激活的情况下,SIRT1仍可被激活;但显然这两条通路可相互协调,以强化对方的效应。
除了调节肝脏和肌肉胰岛素稳态和葡萄糖稳态,SIRT1还可直接作用于胰腺β细胞。SIRT1是胰岛素分泌的正调节因子,可诱发葡萄糖摄入和利用。采用胰腺β细胞特异性过表达SIRT1的小鼠模型(BESTO小鼠)进行体外研究发现,SIRT1水平升高可增加葡萄糖刺激的胰岛素分泌[145];而此种小鼠体内研究也表现为糖耐量改善。与BESTO小鼠研究一致,全身SIRT1缺失小鼠的离体胰岛细胞研究亦显示,其胰岛素分泌功能受损[146]。此两种模型均显示,SIRT1可通过抑制解偶联蛋白2(UCP2)表达以促进胰岛素分泌,而UCP2可升高胞浆ATP/ADP比值。虽然两项研究均显示SIRT1水平可调节胰岛素分泌,然而衰老BESTO小鼠研究显示,单独升高SIRT1水平尚不足以促进胰岛素分泌。BESTO小鼠胰岛素分泌呈增龄性降低,但通过补充NMN后即可恢复。并且随后的研究发现,胰岛细胞SIRT1活性增龄性降低,可能与NAD+生物合成减少有关[147]。这一发现提示,研究NAD+生物合成(而非仅sirtuin)在其他代谢组织中的增龄性变化,具有更为重要的意义。
SIRT1还可调节白色脂肪组织(WAT),这一点具有重要意义,因为衰老进程中脂肪聚集和分布的变化渐趋明显。WAT不仅有储存脂肪的功能,还可作为内分泌器官分泌激素,如瘦素(leptin)、脂联素(adiponectin)和炎症因子[如肿瘤坏死因子α(TNFα)和抵抗素(resistin)]。肥胖还可增加罹患代谢综合征和胰岛素抵抗的风险,而通过CR减轻WAT重量被认为是延长寿命的部分原因。在培养细胞可见SIRT1与PPARγ结合,并抑制与脂肪储存有关的SIRT1靶基因的转录[148]。结果是已分化脂肪细胞SIRT1上调导致脂肪分解,而脂肪储存减少。与上述研究相一致,高脂饮食小鼠给予白藜芦醇或SRT1720后,其体重增幅降低[94,96]。因此,禁食期间会发生脂肪酸动员,而CR期间出现WAT减轻,这些代谢变化的机制可能部分源于SIRT1水平升高。通过加强FOXO1与CCAAT增强子结合蛋白(C/EBP)的相互作用,SIRT1还可促进WAT生成脂联素[149]。因为脂联素可改善胰岛素敏感性,SIRT1对脂联素的调节就成为调节代谢稳态的另一种机制。
|
图5 线粒体sirtuin(SIRT3-5)网络 |
线粒体可代谢能源物质,如脂肪酸、氨基酸和源自葡萄糖的丙酮酸。电子通过电子传递复合物(Ⅰ-Ⅳ)进行传递,从而形成质子梯度(proton gradient),后者再驱动ATP合酶生成ATP。SIRT3可与复合物Ⅰ结合,调节后者活性和细胞能量水平。SIRT3还可与乙酰CoA合成酶2(AceCS2)和谷氨酸脱氢酶(GDH)结合并使其去乙酰化,从而激活其酶活性。SIRT4可通过ADP-核糖基化(ribosylation)结合并抑制GDH活性。SIRT5可去乙酰化并激活氨甲酰磷酸合成酶1(CPS1),此为尿素循环的限速步骤。 |

代谢过程中线粒体Sirtuin的作用
Sirtuin家族中SIRT3–5定位于代谢研究的热点——线粒体[21,35,42,55]。在细胞能量(ATP)生成、ROS生成和凋亡信号转导等方面,线粒体均处于核心地位。线粒体85%~95%的耗氧量用于一系列的细胞酶促反应,最终经氧化磷酸化生成ATP。有能量需求时,线粒体生物合成常伴能源物质的氧化利用增加,以促进能量生成。线粒体可以说是营养物适应性调节的汇聚点(图5):源自葡萄糖的丙酮酸经三羧酸(TCA)循环代谢;脂肪酸和氨基酸分别经线粒体脂肪酸氧化反应和氨基转移酶反应得以转化。不出意料,线粒体功能受损正与衰老相关,并可导致代谢稳态失衡[150]。例如,胰腺线粒体功能损害可抑制ATP/ADP比值升高(其比值升高可刺激胰岛素分泌),而肌肉线粒体功能损害则导致胰岛素抵抗[151]。
近期研究显示,线粒体sirtuin是线粒体变化的核心要素,而只有线粒体发生变化才能促进能量的适应性调节。然而与SIRT1相比,对于SIRT3-5的生化和生物学功能所知尚少。与SIRT1一样,SIRT3也是线粒体sirtuin中最保守的蛋白,对其了解也最为充分。SIRT3可在所有组织中表达,在代谢活跃组织(如棕色脂肪组织、肌肉、肝、肾、心和脑)中水平最高[64,152,153]。
通过实验比较线粒体的膜结合片段和膜可溶性片段,提示SIRT3位于线粒体基质和线粒体内膜。电镜研究发现SIRT3还见于线粒体嵴,此为氧化磷酸化的发生部位。SIRT3缺乏小鼠未见明显的发育或代谢表型[154],且身体成分和线粒体蛋白水平正常,对于禁食和寒冷暴露的反应也很典型,但有一项突出的表型——线粒体乙酰化水平增强[154]。这项重要研究采用质谱方法分析乙酰化程度,发现该型小鼠的代谢相关蛋白(如TCA循环酶类、脂肪酸氧化酶类,以及氧化磷酸化复合体的亚基)均被强烈乙酰化。然而仍需明确,广泛的乙酰化如何影响线粒体的代谢调节。值得注意的是,尚未发现线粒体乙酰转移酶。
SIRT3可结合并去乙酰化至少三种线粒体代谢底物:乙酰CoA合成酶(AceCS)[65,155]、谷氨酸脱氢酶(GDH)和复合物Ⅰ。SIRT3介导的去乙酰化效应似乎可激活酶活性。例如,两个研究小组[154,156]报告,SIRT3可去乙酰化并激活AceCS,后者催化乙酸、CoA和ATP以合成乙酰CoA。正常情况下,哺乳动物细胞并不需要AceCS来合成乙酰CoA。而在生酮情况下(如长期禁食、糖尿病),AceCS活性就显得相当重要;此时耗能组织(如肌肉)需将肝脏释放的乙酸转化为乙酰CoA,以供进一步代谢。SIRT3还可结合并去乙酰化GDH,不过对其活性的效应尚无报道[154,156]。GDH可与谷氨酸互换,将后者转化为α-酮戊二酸;而SIRT3对GDH的调节可能为调节氨基酸流出TCA循环的途径之一。禁食和CR期间,肝GDH活性同样增加,然而对于SIRT3在该反应中所起的作用,尚无相关研究。在体外研究发现SIRT3可调节AceCS和GDH的基础上,有必要进一步明确SIRT3在体内是否调节乙酸或氨基酸代谢。上述相互作用现象提示,在能量限制情况下,SIRT3可将替代碳源(即酮体和氨基酸)汇聚到TCA循环的代谢中心。与这一假说相一致,CR期间棕色脂肪组织和WAT中SIRT3表达增加,而遗传性肥胖小鼠SIRT3表达则降低[153]。
除了可能调节线粒体代谢的核心通路,SIRT3还与线粒体呼吸有关。SIRT3可结合复合体Ⅰ,从而促进NADH驱动的线粒体呼吸,且SIRT3缺失的肝线粒体耗氧量降低,心、肾、肝的ATP基础水平降低50%[157]。过表达SIRT3的棕色脂肪组织耗氧量增加,提示电子传递活性和解偶联增强;相应地,还可见细胞的膜电位降低,ROS生成减少[153]。但是这些过表达SIRT3研究难于解释某些现象,因为其采用的是截短型SIRT3,而其并非恰好定位于线粒体[158]。
鉴于SIRT3、代谢与线粒体功能存在关联,遗传研究又将SIRT3与人类寿命联系起来,这便进一步引发关注。sirtuin保守性核心结构域的沉默性G-T颠换(transversion)与老年男性长寿有关;而SIRT3含有数量不一的串联重复序列,其多态性几乎仅见于90岁以上男性[159,160]。通过深入的生化研究,探索SIRT3单核苷酸多态性(SNP)调节蛋白水平或活性的机制,将成为理解上述相关性的关键。虽然上述人类遗传学研究的范围有限,但其结果提示SIRT3可能对人类寿命有正面影响。
SIRT4定位于人类和小鼠细胞的线粒体,并已在线粒体基质发现[21,22,161]。SIRT4是广泛表达的基因,在小鼠的肾、心、脑、肝和胰腺β细胞中水平最高[21,22,161]。与SIRT3不同,未见SIRT4针对典型的sirtuin靶点显示NAD+依赖性去乙酰化活性[21,22,161]。原因可能是SIRT4底物特异性强于SIRT3,未来可望发现其去乙酰化的特异性底物。不过SIRT4显示具有NAD+依赖性ADP-核糖转移酶活性;现已发现了一种SIRT4底物,SIRT4可与GDH相互作用,并通过ADP-核糖基化(ribosylation)作用抑制后者活性[21]。GDH可调节能量生成过程中的氨基酸利用;其生物学意义明显见于GDH激活突变患者,表现为高胰岛素/高血氨综合征[162]。SIRT4缺乏小鼠的离体胰岛细胞对葡萄糖和氨基酸的反应表现为,GDH活性升高且胰岛素分泌增加。而在另一项独立研究中,过表达SIRT4的胰岛素瘤细胞可抑制胰岛素分泌[161]。SIRT4还显示可与胰岛素降解酶(IDE)相互作用,但这一作用的意义尚不清楚[161]。未来研究中的重要领域中,需要明确SIRT4是否还会影响其他组织的能源物质利用;以及在营养限制或代谢紊乱时,SIRT4如何传递生理变化。
近期,关于SIRT5的第一项综合性研究显示,其可调节氨进入尿素循环。SIRT5定位于线粒体基质,且广泛表达,仅具有微弱的NAD+依赖性去乙酰化作用。SIRT5缺乏小鼠已经产生,其发育正常,未见明显的代谢缺陷[63,154]。SIRT5在细胞代谢过程中至少与两种蛋白相互作用,即细胞色素c和氨甲酰磷酸合成酶1(carbamoylphosphate synthetase 1,CPS1)。CPS1是尿素循环的第一个限速步骤,清除氨基酸代谢所产生的氨有赖于CPS1的活性。SIRT5通过将CPS1去乙酰化以激活其酶活性[63]。SIRT5缺乏小鼠长期禁食可见氨水平升高,也可提示SIRT5可辅助肝脏处理氨基酸代谢的副产物[63]。而SIRT5缺乏是否会增加对氨毒性的易感性,尚没有定论。
人们对线粒体sirtuin调节的研究兴趣日渐浓厚,相关的调节机制包括三个方面:第一,由于Nampt增加,禁食大鼠肝线粒体NAD+水平可升高2倍,同时应激耐受性增强[44,163]。第二,线粒体sirtuin还可在蛋白水平被调节,例如CR、禁食、应激和运动期间,SIRT3水平升高[153,163,164]。第三,线粒体sirtuin的定位也可被调节。例如,细胞应激情况下,SIRT3可由细胞核转位至线粒体[67,68];而由于刺激物不同,SIRT5可主要定位于线粒体的膜间隙或基质[156]。
线粒体sirtuin精巧的代谢调节机制日渐明晰。适应性调节过程中,SIRT3-5之间相互配合,共同调节线粒体代谢的关键步骤。例如,SIRT3和SIRT4均可调节GDH,并刺激氨生成。相应地,SIRT5则通过激活尿素循环,帮助将这些氨清除。未来还需进一步填补有关线粒体sirtuin调节代谢的诸多研究空白:SIRT4可抑制GDH,而SIRT3对后者的效应则未明;被激活的SIRT3和SIRT4是否同时发挥作用?总之,上述sirtuin作用于线粒体代谢的关键环节,可通过促进能源物质进入TCA循环或调节氧化磷酸化而实现。
关于sirtuin在肿瘤生成和癌细胞增殖中的作用,正引起相当大的争论。其中针对SIRT1的令人困惑之处在于,其在细胞存活和细胞死亡方面扮演双重角色,不同刺激情况下被调节到不同方向(图6)。虽然CR被认为是啮齿类和灵长类预防癌症的最有效途径,且某些迹象提示sirtuin为肿瘤抑制因子[70],然而部分sirtuin(如SIRT1和SIRT3)却具有促存活(prosurvival)作用,可能会促进肿瘤生成[165,166]。
最初多数证据支持SIRT1为致癌基因,起始于SIRT1首个底物(肿瘤抑制因子p53)的发现[167,168]。SIRT1可将p53的赖氨酸382去乙酰化,从而降低其反式激活(transactivation)效应,使得细胞绕过p53介导的凋亡通路。这些早期数据以及其他研究,均显示SIRT1可抑制凋亡[88],提示SIRT1可能具有致癌作用。而近期研究却显示,SIRT1在DNA断裂修复中发挥重要作用,并可预防癌症小鼠模型肿瘤发生。具有额外SIRT1拷贝的小鼠未见早死或肿瘤生成增加;实际上,SIRT1转基因小鼠的白血病和结肠癌模型,寿命反而较长[169-172]。由此上述发现引发了相当大的争论,SIRT1在癌症发生过程中发挥的作用是因其被激活还是被抑制[171]。
如何解释SIRT1在癌症中自相矛盾的数据?这可能要深入到对肿瘤遗传学和肿瘤生成阶段的评估。评估sirtuin对肿瘤及其治疗的潜在作用时,须注意的是目前的多数资料均来自细胞系,而这可能会造成误导,从而将结论外推到体内状态。还需意识到,SIRT1研究采用的某些肿瘤(如p53淋巴瘤)模型,更多与肿瘤预防相关,而非肿瘤治疗。原因之一在于,在重要细胞类型中,其SIRT1可能位于促细胞存活(而非致癌)的反馈环中。这一假设是否成立,实际情况又是如何?这些方面仍是未来SIRT1研究的重点,并且更多需要的不仅是细胞培养研究,还要有体内研究。
|
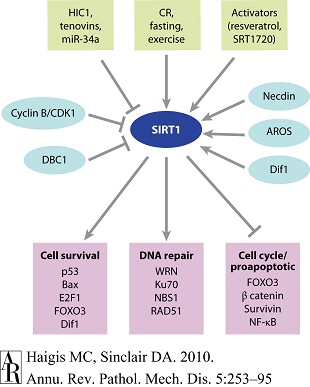
图6 SIRT1针对细胞存活和凋亡发挥的重要作用 |
鉴于对SIRT1的调节相当复杂,欲揭示其在肿瘤中的作用也颇有难度。调节SIRT1基因的因素包括环境刺激(如禁食、运动)、微RNA(miRNA)、tenovin,以及肿瘤高度甲基化蛋白1(HIC1)转录阻遏因子。SIRT1还可通过与以下蛋白相互作用而受到调节,即乳腺癌缺失蛋白1(DBC1)、SIRT1主动调节剂(AROS)、Dif1和necdin。SIRT1可与某些蛋白(涉及到细胞存活、DNA修复和凋亡)相互作用并使其去乙酰化(图3)。基于上述信息,仍难以预测过表达SIRT1或SIRT1激活剂的体内效应,不过目前的相关实验研究[171]提示SIRT1可作为肿瘤抑制因子,这些研究分别采用p53−/+淋巴瘤模型、乳腺癌易感基因1(BRCA1)乳腺癌模型、二甲基苯并蒽(DMBA)皮肤癌模型和APCmin+/−结肠癌模型。 |
缩写 CR:热量限制;DBC1:乳腺癌缺失蛋白1;FOXO:叉头框蛋白O;HIC1:肿瘤高度甲基化蛋白1;NF-κB:核因子-κB。 |
肿瘤中sirtuin表达的变化
目前有关sirtuin在肿瘤中表达水平的信息绝大多数来自SIRT1,其次来自SIRT2。与正常组织比较,SIRT1在多种类型的肿瘤中表达水平较高,其中包括结肠癌、皮肤癌、乳腺癌、前列腺癌和白血病[121,173-177]。不过这种相关性并不明确。Wang等[178]通过分析公用数据库发现,前列腺癌、膀胱癌、胶质母细胞瘤和卵巢癌SIRT1 mRNA水平较正常组织降低。他们发现BRCA突变肿瘤患者SIRT1 mRNA水平显著低于野生型BRCA1肿瘤患者。对45名乳腺癌患者样本进行免疫组化染色也发现,BRCA1与SIRT1表达之间存在正相关。
近期的一项胰腺癌研究发现,11例患者中仅有1例SIRT1水平升高[179];而其他sirtuin与肿瘤组织更具相关性,异常发现分别为SIRT2 6例、SIRT3 4例、SIRT64例,其中最显著的发现是,这11例胰腺癌中有10例SIRT5转录产物升高。处于0期、ⅠB期、ⅡB期和Ⅳ期的全部患者,均可见SIRT5 mRNA水平升高,一例Ⅲ期患者SIRT5下调。
鉴于SIRT2在应激反应中可发挥调节细胞周期的作用,故而有提示其在肿瘤生成中可能具有某些作用,可能部分应用于化疗。新近证据提示,SIRT2可解除严重损伤细胞的有丝分裂停滞,从而使其进入凋亡阶段[17,180,181]。在神经胶质瘤和神经胶质瘤衍生的细胞系,可见SIRT2表达普遍降低(通过修饰SIRT2启动子组蛋白或删除SIRT2等手段发现)。因此,抑制SIRT2可能使细胞倾向于失控性生长[182]。与此假说相一致,近期研究发现组蛋白H3赖氨酸56(H3K56,为SIRT1/SIRT2靶点之一)在癌组织中高度磷酸化;并且,无致癌性的MCF10A细胞乙酰化H3K56水平,显著低于有致癌性的MCF7乳腺癌细胞[183]。
总之,上述数据提示SIRT1/SIRT2活性降低与肿瘤生成有关,而在某些癌症中提高SIRT2活性可能是有益的;细胞生物学实验亦支持这一观点。异位表达野生型SIRT2可延缓细胞周期进程,并增加多核细胞数量[60,180]。CDK1可将SIRT2丝氨酸368磷酸化,从而抑制细胞生长[60,180]。作为对微管抑制剂[如诺考达唑(Nocodazole)]的反应,过表达SIRT2可使细胞周期停滞于有丝分裂之前[181,184]。另一方面,抑制SIRT2可促进中心体分裂,延迟发生诺考达唑所致的慢性有丝分裂停滞,但也可防止重新进入细胞周期的细胞死亡[181]。有趣的是,SIRT2催化突变体(catalytic mutant)也可使多核细胞数量增加[60,180],提示实现精确的有丝分裂需要严格的SIRT2水平,而这又依赖于具体环境;所以SIRT2抑制剂或激活剂治疗某些肿瘤可能有效。
与非恶性乳腺细胞相比,乳腺癌细胞SIRT3 mRNA和SIRT7mRNA水平均增高,且与淋巴结阳性相关[185]。某项研究报告在甲状腺癌中“SIRT8”表达增加,后证实应为SIRT7表达增加(注意:并不存在SIRT8)[186]。总之,各种sirtuin与特定肿瘤均无明确的相关性;可能的例外是发现BRCA1阴性乳腺癌缺乏SIRT1,该研究调查其它sirtuin与肿瘤可能的相关性,但由于样本量太小,尚无法作出有意义的结论。
SIRT1作为潜在的致癌基因
大量的细胞培养实验数据显示,SIRT1可预防细胞凋亡和衰老[62,128,187,188],提示体内抑制SIRT1可能对某些肿瘤类型有效[128,165,189]。2001年发现,SIRT1可靶向p53,并与之相互作用,使其去乙酰化,从而降低p53与DNA的结合能力[102,168,190]。表达额外SIRT1的细胞为应答DNA损伤和细胞应激,可表现为典型的乙酰化p53赖氨酸382水平降低;且此种细胞通常可耐受p53依赖性细胞周期停滞和凋亡[167,168]。与这一发现相一致,表达H363Y显性失活(dominant-negative)SIRT1等位基因的细胞,或通过小干扰RNA(siRNA)沉默SIRT1 mRNA,均可增加p53依赖性转录活性[167],并增强细胞对应激的敏感性[171,191]。类似地,取自SIRT1敲除小鼠的细胞,或给予siRNA以对抗SIRT1的细胞,均显示p53赖氨酸382和320高度乙酰化[188,192]。sirtuin抑制剂sirtinol可诱导衰老样生长停滞,并可致人乳腺癌MCF7细胞和肺癌H1299细胞中纤溶酶原激活物抑制因子1(PAI1)表达降低[193]。为应答表皮生长因子(EGF)和胰岛素样生长因子-1(IGF-1),细胞生长停滞的同时还伴见对c-Jun氨基端激酶(JNK)和p38丝裂原激活蛋白激酶(MAPK)的激活受损,且Ras亦减少。而经sirtinol处理后,EGF受体和IGF-1受体的酪氨酸磷酸化,以及对蛋白激酶B的激活则不受影响。该小组[194]研究内皮细胞功能时还发现,抑制SIRT1可增强p53磷酸化,并诱导早衰样表型,而过表达SIRT1则可防止出现衰老样改变。但有其它研究显示,通过siRNA特异性抑制SIRT1可促进肿瘤细胞死亡,而对培养的正常细胞并无毒性效应[188]。该实验中细胞死亡的机制仍不清楚,因为p53在SIRT1损耗所致的癌细胞杀伤效应中并非必需,当然也不能排除p53的参与[188]。
人成纤维细胞SIRT1表达增加可促进细胞增殖,减少细胞衰老,并使细胞生长率升高;同时还可使人胚肺二倍体成纤维细胞(2BS)的寿命延长,p16INK4A表达减少,并促进视网膜母细胞瘤(Rb)蛋白磷酸化[195]。该实验研究者[195]还提出激活细胞外信号调节激酶/核糖体蛋白S6激酶1(ERK/S6K1)可诱导衰老,通过过表达SIRT1或给予SIRT1激活剂白藜芦醇即可引发。过表达SIRT1还可抑制肿瘤抑制因子和DNA修复基因[包括FOXO1、FOXO2、FOXO4、沃纳综合征蛋白(WRN)、Rb、p73、错配修复基因1(MLH1)和奈梅亨断裂综合征蛋白1(NBS1)]的表达或活性[171,196]。近期斑马鱼和小鼠研究发现,SIRT1还可促进血管生成[197,198],也提示SIRT1具有促癌作用。
Baylin和同事研究[172,199]表明,SIRT1介导的上皮型钙粘蛋白(E-cadherin)沉默在肿瘤生成中发挥一定作用。上皮细胞癌中E-cadherin基因的CpG岛因高度甲基化而频繁沉默。如果DNA断裂首发于CpG岛,则DNA甲基转移酶3B(Dnmt3B)的短暂募集,以及随后的DNA甲基化(使DNA沉默)都需要SIRT1参与。该结果与近期的若干研究相一致,均认为SIRT1与断裂DNA的修复有关[170,172,178,200],而体内SIRT1是否可通过这一机制促进上皮细胞癌生成,尚有待研究。由此SIRT1抑制剂对此类肿瘤的功效引发关注,特别是需明确DNA甲基化所致沉默是否可逆。
两种肿瘤抑制因子已被鉴定为SIRT1负调节因子。近期研究显示,肿瘤高度甲基化蛋白1(HIC1,为具有锌指/BTB结构域的蛋白,受p53调节)可与SIRT1结合,随后即抑制SIRT1基因转录[102]。而失活HIC1则上调SIRT1转录,继而使p53失活,而DNA受损的细胞则得以绕过凋亡通路。重要的是,研究者发现HIC1启动子会发生增龄性过度甲基化,从而导致SIRT1增龄性上调,最终增加肿瘤易感性。值得注意的是,抑制糖酵解可减少辅阻遏因子(corepressor)E1A羧基端结合蛋白(CtBP)与HIC1的结合,增加SIRT1表达,从而将氧化还原状态与SIRT1表达联系起来[103]。
另一种负调节SIRT1的肿瘤抑制因子为DBC1[128,129,201],其可与SIRT1的氨基端形成稳定的相互作用,从而抑制SIRT1的去乙酰化活性。通过siRNA抑制DBC1可促进p53的去乙酰化,使细胞在遗传毒性应激下存活(该效应依赖于SIRT1完成)。这些研究数据表明,DBC1缺失促发乳腺癌的部分原因在于激活了SIRT1,进而下调p53和(或)其他肿瘤抑制通路。
SIRT1作为潜在的抑癌基因
虽然细胞研究已提示SIRT1可能为促癌基因,但近期研究却发现SIRT1还可作为抑癌基因。首先,催化失活H363Y等位基因以抑制SIRT1基因,或特异性抑制细胞SIRT1,均不影响细胞存活或细胞生长,也不足以诱导对内源性p53的激活[110,188,195]。Solomon等研究发现[110],尽管有DNA损伤和p53乙酰化增强,但细胞死亡并未增加。其次,Mayo和同事的细胞培养研究显示[202],SIRT1可促进TNFα诱导的细胞死亡,说明其还可促进凋亡,而非仅抑制凋亡。
近期研究强调,某一重要的反馈环能够解释体内SIRT1如何抑制肿瘤生成[109]。已知c-Myc基因编码一种原癌基因转录因子,其可调节细胞增殖、细胞生长、凋亡和干细胞自我更新。c-Myc可与SIRT1启动子结合,并诱导SIRT1表达;而SIRT1随后会与c-Myc相互作用,并使后者去乙酰化,导致c-Myc稳定性降低。这一c-Myc–SIRT1反馈环能够阻止细胞性状转化,正符合SIRT1在肿瘤抑制过程中扮演的角色。
在第一项验证SIRT1促进细胞存活或死亡(即促癌活性或抑癌活性)的动物实验中,通过小鼠结肠癌模型(APCmin/+)过表达SIRT1[169]发现,野生型min基因剩余拷贝的缺失可致β-连环蛋白(β-catenin)重定位于细胞核,从而激活转录基因(如myc、cyclin D1),进而激活细胞周期。已知CR可减少肿瘤发生[203],而白藜芦醇亦有类似作用[204,205],但尚不明确过表达SIRT1是否增加或减少肿瘤发生。小肠和结肠过表达SIRT1的小鼠可见腺瘤的体积和数量减少3/4。Ki67和TUNEL染色显示,过表达SIRT1的小鼠肿瘤生长缓慢,凋亡增加。而SIRT1去乙酰化β-连环蛋白可能是这一过程的作用机制。总之,上述研究数据表明,激活SIRT1并由β-连环蛋白驱动,可能对结肠癌及其他肿瘤有潜在治疗价值。
p53杂合体小鼠广泛用于研究小鼠的基因组不稳定性[206]。暴露于电离辐射的p53+/-小鼠可见肿瘤发生增多,原因即在于p53基因座杂合性丧失(LOH)增多[207]。考虑到细胞培养实验中SIRT1可下调p53,激活SIRT1可能会恶化动物的p53+/-表型,并缩短其寿命;然而事实恰恰相反。一项研究中发现,给予实验动物白藜芦醇可增加其存活24%,减少致命性胸腺淋巴瘤发生约45%。SIRT1可防止辐射诱导的肿瘤发生,而未经辐射的p53+/-小鼠的整体肿瘤谱也与前者高度一致[206,208]。进一步使p53+/-小鼠胸腺细胞过表达SIRT1,经辐射后其存活率较对照动物高约46%,而MISTO小鼠致命性胸腺淋巴瘤的发生率降低45%。
补充性研究中,将SIRT1缺失小鼠与相同的p53+/-品系杂交[109]。由于此前细胞培养数据已显示SIRT1可下调p53,所以期待SIRT1−/−与p53+/−联合将会延迟小鼠肿瘤生成(较之单纯p53−/+小鼠)。但对比发现,SIRT1−/−小鼠肿瘤生成进程加速,约5月龄时即有多种自发性肿瘤生成;20月龄时,76%的SIRT1−/−p53+/−小鼠发生肿瘤,而对照组约为10%-15%。原发肿瘤染色体核型分析(karyotyping)显示,存在异倍体(aneuploidy)和染色体畸变(chromosomalaberrations)[178]。两项独立研究证实,SIRT1激活剂白藜芦醇可延迟该品系小鼠肿瘤发生,并延长其寿命[170,178]。并且在衰老过程中,DNA损伤可致SIRT1重新定位,导致启动区SIRT1缺失,并改变基因的表达[170]。上述研究数据支持了衰老的染色质修饰物重定位(relocalizationof chromatin modifiers,RCM)假说,该假说认为DNA损伤促使染色质修饰蛋白重新定位,造成基因表达发生变化,从而导致衰老[170,209]。然而还需要更多的研究来验证这一模型(图7)[196]。
近交系背景的SIRT1−/−小鼠具有胚胎致死性(embryonic lethality)[129]。基于细胞培养数据显示,SIRT1可失活p53,最初推测上述胚胎致死性源于p53的高度激活;由此预测,删除p53的SIRT1敲除小鼠可能会避免前者出现的致死性,然而事实并非如此[178,210]。在题目为“SIRT1fails to affect p53-mediated biological functions”(SIRT1不影响p53介导的生物学功能)的文章中,McBurney和同事[210]通过分析SIRT1敲除小鼠胚胎组织,并未发现p53下游基因的变化。
|
图7 衰老的染色质修饰物重定位(relocalization of chromatin modifiers,RCM)假说 |
该假说由S. Imai和H. Kitano于1998年提出,认为异染色质(heterochromatin)的变化是衰老过程的深层原因。这一观点部分基于酵母研究,其显示为应答DNA损伤和衰老,酵母SIR2可从沉默基因座释放出,并重新定位于DNA断裂处;推测这一过程可重组染色质,从而易化修复过程。重定位过程中,沉默性交配型基因(HML和HMR)的表达可导致不育(衰老特征之一)。近期工作显示在哺乳动物中亦存在类似的促进衰老机制。为应答DNA断裂或衰老,SIRT1也可从开放阅读框(ORF)重定位于DNA断裂区,并似可改变断裂区周围的染色质,从而募集DNA损害修复蛋白(如RAD51和NBS1)。SIRT1的重定位,或其诱导的表观遗传学变化,可能改变基因表达的模式,从而导致组织功能异常和增龄性疾病。 |
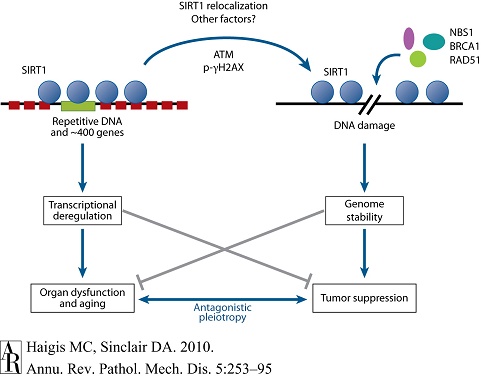
支持SIRT1具有肿瘤抑制作用的证据是,SIRT1基因在修复断裂DNA和保持基因组稳定性方面扮演重要角色。1999年发现了第一条线索,3家独立实验室的研究显示,酿酒酵母为应答DNA损伤,将其SIR2(sirtuin家族的早期成员)定位于断裂的DNA[211-213]。这一反应只要一条DNA断裂即可引发,并通过RAD9和MEC1(酵母为ATR/ATM)实现检查点(check-point)的信号转导。而在当时尚不清楚,作为去乙酰化酶的SIR2如何参与断裂DNA的修复,更不必说其他的sirtuin[214]。后来发现SIR2、SIR3和SIR4蛋白均可使未修复的断裂DNA绕开G2期停滞[215]。Tyler和同事[216]研究发现,在DNA双链断裂发生约3h后,SIR2即结合于断裂修复位点,同时组蛋白乙酰化程度降低。上述数据表明,SIR2可在DNA断裂位点周围进行染色质的修饰,从而易化DNA修复过程[216]。
四年后,系列研究论文显示,哺乳动物SIRT1可调节MRE11/RAD50/NBS1复合体中的重要修复因子NBS1[217]。之后几个月有报告说,SIRT1可定位于DNA断裂处,且断裂DNA的有效修复需要SIRT的参与[170,178]。与上述结论相一致,SIRT1缺乏小鼠更易出现DNA损伤诱导的异倍体,断裂DNA的修复效率降低约50%[170]。为应答ATM和H2AX介导的信号(包括NBS1和RAD51),缺乏SIRT1的细胞募集DNA修复因子到达断裂处的过程存在缺陷。这些数据或可解释以下现象,即过表达SIRT1小鼠不易发生p53基因座杂合性丧失和肿瘤;而SIRT-/-小鼠则正好相反[171]。这些数据还可部分解释这一现象,即SIRT1可抑制APCmin+/−小鼠腺瘤的发生(该腺瘤由min基因座杂合性丧失所致)。但以上并不能完全解释腺瘤发生减少的原因,因为过表达SIRT1还可增强凋亡,并减少瘤体中有丝分裂细胞的数量[169]。
1997年首先有报道,白藜芦醇可保护小鼠免受化学诱导性皮肤癌的侵袭[218]。此后,众多文献证实,白藜芦醇可作为肿瘤小鼠模型的强效化学保护剂。有趣的是,McBurney实验室近期发现[125],白藜芦醇对SIRT1缺陷小鼠的抗皮肤癌效应显著降低。给予白藜芦醇的野生型小鼠中仅20%在15周后发生肿瘤,而SIRT1缺陷小鼠中则为75%。因此,该模型中白藜芦醇的抗皮肤癌效应部分由SIRT1介导。
考虑到酵母Sir2对端粒有重要作用,许多研究者推测SIRT1可能也与端粒的生物学功能有关。SIRT1并未见于哺乳动物的端粒区,然而取自SIRT缺陷小鼠的造血干细胞生长能力增强,似乎与端粒酶活性增强有关[219]。该项研究中,单独过表达或下调SIRT1,对人二倍体成纤维细胞的寿命均无影响,提示SIRT1敲除小鼠在发育过程中可能发生了遗传学或表观遗传学的改变。不过,过表达SIRT1细胞系的生长速度较慢[219]。
肿瘤中其他sirtuin的作用
已知具有肿瘤背景的sirtuin还有SIRT2,是一种微管蛋白去乙酰化酶,为正常有丝分裂过程所必需[17,180],并可调节有丝分裂前中期的检查点功能,以防止染色体不稳定情况的发生。有丝分裂期间,SIRT2大量增加,且在细胞周期由G2期转换到M期的过程中,SIRT2被多重磷酸化。稳定过表达野生型SIRT2的细胞(而非缺乏SIRT2活性的错义突变细胞)的有丝分裂期明显延长[220]。神经胶质瘤和神经胶质瘤衍生细胞系中,SIRT2的表达下调,提示SIRT2表达增加可能对肿瘤有益。
体外发现,抑制或下调SIRT2可干扰细胞周期进程,并促使细胞周期停滞[180],提示抑制SIRT2可能为治疗某些肿瘤的有效方法。一种新型SIRT2抑制剂AC-93253(可选择性抑制SIRT2,而非SIRT1)显示,对四种不同的肿瘤细胞系均具有细胞毒性[221]。而SIRT1和SIRT2双重抑制剂cambinol则显示,可抑制伯基特淋巴瘤(Burkitt lymphoma)异种移植物(xenograft)(图2)[222];不过由于脱靶效应,cambinol是否通过SIRT1或SIRT2起效尚未明确。一条可能的线索来自近期的一篇报道,其发现SIRT1和SIRT2均可去乙酰化H3K56,而后者作为去乙酰化标志物在多种类型肿瘤中均增加[183]。很显然,上述化合物是否能够成为临床有效的肿瘤治疗药物,还有更多的工作要做。
而对于SIRT3-7等其他sirtuin,仅有少量线索提示其在肿瘤生物学中有重要作用。在HCT116细胞中,SIRT3通过JNK2(独立于SIRT1的通路)发挥促凋亡作用[223]。而在其他情况下(如应答线粒体NAD+水平低于临界值时的DNA损伤),SIRT3和SIRT4则具有抗凋亡效应[44]。已知15号染色体的GCIP(或CCNDBP1/DIP/HHM)为潜在肿瘤抑制因子,在结肠癌、乳腺癌和前列腺癌中表达下调。GCIP可与SIRT6(为HDAC蛋白Ⅲ亚型)特异性相互作用,为保持基因组稳定发挥重要作用[22,224]。这表明,GCIP-SIRT6有可能具有肿瘤抑制作用[225]。
总之,尽管研究者试图通过小鼠模型检验SIRT1的作用,但并无确凿的体外研究数据表明SIRT1为促癌基因。过表达SIRT1的所有品系小鼠中(至少有6种模型已有报道),均显示无肿瘤易感性,即使对于肿瘤易感小鼠模型(如p53+/−和APCmin/+)亦是如此。而针对SIRT1缺失小鼠也无强有力证据以证实SIRT1为肿瘤抑制基因,实际上此种小鼠更易发p53诱导性肿瘤。造成细胞培养研究与体内研究差异的可能原因,一是由于SIRT1只针对特定类型细胞和(或)特定小鼠模型细胞;二是SIRT1的体内作用并非促进细胞生长,而是防止因细胞早衰导致衰老和疾病;三是SIRT1可与p53相互作用,从而改变线粒体呼吸或线粒体其他功能[226,227]。只有针对人类组织样本和动物模型进行更多的研究,我们才能够深入理解是否SIRT1能够促进体内肿瘤生成和生长(若是,则在何种情况下促进)。
心血管疾病(CVD)作为造成死亡的首要原因,是一个进行性、多阶段的过程;该过程取决于胆固醇生物合成、免疫系统和血管内皮细胞功能之间复杂的相互作用[228]。CVD的危险因素包括氧化低密度脂蛋白(ox-LDL)胆固醇升高、血管损伤,以及炎症程度加重和胆固醇沉积(因巨噬细胞浸润到血管内皮下层的平滑肌所致)。数十年来,动物研究[85]和人类研究[229]已发现,通过CR可预防CVD;然而这一效应是否仅通过遗传通路介导,还是通过药物干预诱发,目前仍不明确。
越来越多的证据提示sirtuin可介导(至少部分介导)CR[230,231],由此有推测认为sirtuin预防或治疗CVD可能有效[5]。近期将SIRT1作用于CVD的研究数据作一汇总后,证实了上述观点,也使得SIRT1有望成为有效治疗和干预CVD的分子靶点[232]。SIRT1在内皮细胞高度表达,对抑制动脉粥样硬化(AS)进展的关键环节也具有调节作用,如上调内皮型NO合酶(eNOS)、减轻平滑肌细胞衰老、抑制动脉炎症和ROS,以及促进血管生长[232,233]。SIRT1还可改变肝脏和巨噬细胞的胆固醇生物合成[143],降低血脂水平[138]。总之,上述结果正与CR预防CVD中SIRT1的重要作用相一致。不过,对于SIRT2-7在CVD中的潜在作用还仅仅是推测。
除酵母SIRT1与CR的联系之外,首先提示SIRT1对哺乳动物CVD具有预防作用的报道,是有关白藜芦醇与SIRT1关系的研究。在1990年代早期,就已将白藜芦醇宣扬为红酒心血管保护效应的根源;而细胞培养和动物模型均显示其具有高度的抗炎和预防CVD作用[234]。直到2003年,这项保护作用的机制仍被全部归因于白藜芦醇的抗氧化特性。2006年发现白藜芦醇对高脂饮食老年小鼠主动脉的ROS、炎症和凋亡均具有抑制作用,且可维持正常的内皮功能[96]。虽然上述效应与去乙酰化的PGC-1α(SIRT1靶点之一)效应一致,但这些研究仍可提示SIRT1涉及到白藜芦醇的保护效应。而此后越来越多的证据认为SIRT1与上述保护效应直接相关。已知针对心血管系统损伤进行重构和修复,血管新生(angiogenesis)是关键因素之一。Potente和同事发现[197],SIRT1缺乏小鼠血管内皮的发育过程看似正常,但应答缺血的血管新生能力受损。对发育的斑马鱼胚胎采用荧光标记其内皮细胞,通过内皮细胞培养并对血管形成进行延时分析(time-lapseanalysis),发现SIRT1是内皮芽生(endothelial sprouting)和血管迁移(vessel navigation)必不可少的因子。而SIRT1敲除成年小鼠则未见发育异常,这是否与sirtuin的丰富性有关,在这方面小鼠与斑马鱼又有何差异,都有待于研究。
SIRT1主要的心血管保护效应为抑制炎症。给予离体巨噬细胞和内皮细胞SIRT1激活剂,可降低炎症细胞因子水平,包括TNFα、细胞间黏附分子-1(ICAM-1)、白介素-6(IL-6)、IL-1和诱导型NOS(iNOS)[121,235,236]。与应答SIRT1激活类似的抗炎反应,可见于高脂饮食和香烟烟雾暴露的小鼠和大鼠[95,237,238]。培养的冠脉内皮细胞(CAEC)中,白藜芦醇的保护效应可因SIRT1基因被抑制而消失,而过表达SIRT1则可模拟白藜芦醇的保护效应[238]。
CR和药物激活SIRT1对心血管的某些益处,可能源于SIRT1可诱导NO信号转导。已知eNOS可生成NO,不仅具有抗AS作用,还可促进血管舒张。白藜芦醇显示可诱导eNOS表达,且单用白藜芦醇即可起效[205,239],也可与某种他汀联合起效[240];不过在某项大鼠研究中其未能诱导eNOS表达[241]。
建立eNOS与SIRT1直接联系的第一项研究发现,SIRT1可与eNOS相互作用,并使后者的赖氨酸496和506去乙酰化[两位点为eNOS的钙调蛋白(calmodulin)结合域],从而导致eNOS活性增强[242]。CR饮食小鼠的eNOS乙酰化水平较低,正与CR啮齿动物SIRT1和eNOS水平升高相一致[88,243,244]。NAD+生物合成基因Nampt/PBEF和SIRT1激活剂,均可使处于高糖环境的人内皮细胞复制寿命延长,并增强其血管新生能力[245]。反之,抑制SIRT1表达则降低NO的生物利用度,抑制内皮依赖性血管舒张,并诱导内皮细胞早衰[242]。与体外研究一致,过表达SIRT1的转基因小鼠内皮细胞可防止血管舒张性丧失,并降低apoE−/− AS模型的AS斑块数量[246],且对血脂、血糖水平无影响[247]。有趣的是,CR诱导SIRT1还需通过eNOS信号转导通路[244],表明SIRT1–eNOS–SIRT1信号转导通路可形成正向反馈环,从而放大CR和白藜芦醇的效应(图8)
|
图8 SIRT1在动脉粥样硬化(AS)和心血管疾病(CVD)中的保护作用 |
AS一般进程为,首先因血管壁损伤和炎症促进巨噬细胞浸润,后者吸收并聚集氧化低密度脂蛋白后成为泡沫细胞。最终泡沫细胞破裂,并促进炎症反应和斑块形成,进而阻塞血流。内皮细胞过表达SIRT1或给予白藜芦醇的小鼠显示,SIRT1可减少血管壁活性氧(ROS)生成并延缓CVD进程。这一效应涉及多种机制,包括通过增加内皮型NO合酶(eNOS)活性,以及降低核因子-κB(NF-κB)活性以抑制炎症反应。而增加巨噬细胞的胆固醇逆向转运,也可能为有益的促进因素。 |
缩写 cGMP:环磷酸鸟苷;ICAM-1:细胞间黏附分子-1;IL-6:白细胞介素-6;iNOS:诱导型NO合酶;TNFα:肿瘤坏死因子α |

近期Ungvari和同事发现[238],给予白藜芦醇可减轻香烟烟雾提取物对大鼠动脉和CAEC的毒性效应;且炎症标志物(ICAM-1、iNOS、IL-6和TNFα)有相当程度的降低,激活的核因子κB(NF-κB)和炎症基因表达也同样降低。CAEC中此种保护效应(包括抑制凋亡)可通过抑制SIRT1基因而被阻止,而过表达SIRT1则可模拟白藜芦醇的保护效应。因此认为,白藜芦醇的血管保护和抗炎效应均需要SIRT1的参与,由此SIRT1成为治疗CVD和血管衰老的潜在药物靶点。
至于其他sirtuin(即SIRT2-7),其对心血管系统的潜在影响所知不多。考虑到SIRT3为应激应答和运动应答基因,可保护心肌细胞抵抗遗传毒性和氧化应激介导的细胞死亡,所以认为SIRT3可能会防止心梗或慢性心衰所致损伤。细胞核型SIRT3则可通过去乙酰化Ku70,使其与线粒体的促凋亡蛋白Bax相隔离,从而促进细胞存活[163]。
血管形成(vascularization)
SIRT1与CVD相关的另一个特征是其可下调FOXO信号。FOXO转录因子家族成员均为必不可少的血管形成负调节因子,而其中FOXO1为首要的中介物[248-250]。SIRT1可使FOXO转录因子去甲基化,从而降低FOXO依赖性转录活性[248]。SIRT1还可与Notch信号转导通路的成分Hey2蛋白相互作用[251],而Hey2属于发状分裂相关增强子(HESR)家族的碱性螺旋-环-螺旋(bHLH)抑制因子(Hairy and Enhancer of split basic loop-helix-loop-helix repressor)。正如Potente和Dimmeler所指出的[198],考虑到Hey2在脊椎动物血管形成模式(vascular patterning)中的作用,SIRT1与Hey2的相互作用提示,SIRT可能在Notch的下游起效,从而调节含Hey2内皮的血管生成活性[198]。
已知ox-LDL胆固醇在AS进程中发挥关键作用,而高密度脂蛋白(HDL)则具有保护作用。Guarente和同事[143]发现,SIRT1可去乙酰化并激活核受体LXR,促进LXR靶基因的转录;涉及的LXR靶基因中有些与脂质代谢相关[包括ATP结合盒转运体A1(ABCA1)],有些则介导胆固醇从外周组织流出。于是引发推测,CR激活SIRT1或应用SIRT1激活剂的长期效应,可能是促进胆固醇从外周组织流出,从而延缓AS斑块的形成[198]。肝脏SIRT1可通过激活某些基因[如固醇调节元件结合蛋白1(SREBP1)]调节肝内甘油三酯(TG)合成。与此发现相一致,SIRT1敲除小鼠血浆TG降低,且肝内TG聚集减轻,同时HDL胆固醇升高;但奇怪的是,糖耐量也增加[143]。
与sirtuin在CVD中的情况不同,其在神经变性病变中的作用并不完全清楚。虽然众多文献显示NAD+和SIRT1增加具有神经保护作用[252,253],关于SIRT1的未知问题仍然存在。是否只有激活SIRT1才有益处,在某些情况下是否应该抑制SIRT1?为何神经保护作用不必完全需要SIRT1催化活性的参与?目前也有争论,白藜芦醇和NAM具有的神经保护作用是否经SIRT1的介导而实现,或者是由于其他的sirtuin,抑或是存在完全不同的机制。甚至有研究认为SIRT1对神经元有害[98]。而对于其他sirtuin(SIRT3-7)则所知甚少。有一篇报道说[254],增加SIRT2、SIRT3和SIRT6的表达可促进暴露于低钾的神经元的凋亡,而SIRT6则可中度保护神经母细胞瘤细胞对抗同型半胱氨酸毒性。还有两项研究报告SIRT3和SIRT4具有细胞保护作用[44,163];其中后一项研究显示,两者的细胞保护作用通过增加Nampt和线粒体NAD+而实现[44]。引发研究兴趣的是,这种保护作用是否在神经元也存在。
在2000年代前期,已有研究显示SIRT1可抑制多种细胞类型的凋亡[31,88,168,187,255],而Milbrandt和同事首先则发现[252],SIRT1还具有神经保护作用。其研究显示,SIRT1是保护神经元对抗华勒变性(WD,其实质为神经挤压伤继发的轴突末梢死亡[252,256])的必要成分。WldS突变小鼠对WD的耐受显著增强的原因在于,该突变可增强NAD+生物合成基因Nmnat与泛素组装蛋白Ufd2的融合[257,258]。Milbrandt在论文中提出[252],Nmnat所致的NAD+生成增加,以及随后SIRT1的激活,正是神经保护作用的必要条件。目前对SIRT1在多种情况下均具有神经保护作用并无异议,且已知SIRT1在人类神经元中需具备催化活性,但以下问题仍悬而未决,即Nmnat1如何保护细胞,这其中SIRT1是否参与[259,260]。举例来讲,现已发现SIRT1在NAD+发挥保护作用过程中并非必需,而研究数据相冲突之处在于,Nmnat在体内是否可单独保护神经元,或其催化活性是否为必备条件[253,259,261-263]。而近期的体内研究数据则可支持Milbrandt提出的观点[253]。最近另一项研究显示,白藜芦醇可抵消WldS小鼠对WD的耐受作用,研究者认为这一现象是由SIRT2被激活所造成的[264];不过考虑到白藜芦醇在体外并不激活SIRT2[94],上述研究结果让人颇感迷惑。并且这项研究似乎还与以下研究背道而驰,其发现抑制SIRT1对帕金森病(PD)小鼠模型具有神经保护作用[265]。
虽然SIRT1和SIRT2在WD中的作用仍不明确,但体外和体内研究显示,SIRT1在多种细胞应激情况下均具有神经保护作用。2004年,Brunet等首先报告[187],采用小脑颗粒神经元研究发现,SIRT1可介导产生神经保护作用;具体机制为SIRT1可靶向FOXO3,并使之去乙酰化,继而抑制FOXO介导的细胞死亡。同年,Sinclair和同事发现[62,88],SIRT1可靶向Ku70羧基端,并使之去乙酰化,从而促进Bax与Ku70结合,这样便可阻止Bax启动凋亡进程。
已知SIRT1和STAC在体外和体内对神经元均具有神经保护作用。Tsai和同事报告[139],SIRT1保护神经元避免凋亡是由于突变SOD1-G37R的过表达,后者为肌萎缩侧索硬化相关等位基因。作者还报告,SIRT1可对抗与细胞周期蛋白依赖性激酶5(CDK5)相关的p25突变,此型突变可能会促发轴突病变和阿尔茨海默病(AD)。在p25转基因小鼠AD和轴突病变模型中,白藜芦醇和SIRT1可预防神经变性和认知衰退[139]。Pasinetti和Sauve研究小组报告[86],采用表达人APPcDNA的瑞典Tg2576小鼠AD模型发现,人SIRT1和白藜芦醇还可降低神经元Aβ含量。文中指出,CR的神经保护作用可能是由于NAD+/NAM比值发生改变,随后经SIRT1介导Rho激酶1(ROCK1)被激活,而ROCK1可诱导具有保护作用且不生成Aβ的α-分泌酶通路[86]。
视神经炎为视神经的炎症病变,通常因视神经髓鞘肿胀和破坏而致失明;本病为多发性硬化的前驱症状。Shindler和同事发现[267],通过玻璃体内注射白藜芦醇或NAD+前体NR以激活SIRT1,可显著减轻视网膜神经节细胞的丧失程度;而这种保护效应可被SIRT1抑制剂sirtinol所阻断。而另一种眼病老年性黄斑变性,则是导致老年人失明的首要原因。补体因子H基因(CFH)多态性可降低CFH活性,增加老年性黄斑变性的发病风险。过表达SIRT1可降低FOXO3被募集到CFH基因调节区的程度,从而逆转H2O2诱导的CFH基因表达被抑;由此,通过激活SIRT1改善或延缓老年性黄斑变性进程的可能性进一步增加[268]。
近期研究显示,SIRT1可控制神经祖细胞(neural progenitor)的命运,而后者被认为可再生神经元以代偿中枢神经系统的损伤。脑部多种神经变性疾病的典型表现为星形胶质细胞增多(较之神经元),这一过程通过反应性星形胶质细胞增生(reactiveastrogliosis)而实现,由炎症所致的氧化反应所诱导。近期研究发现,通过白藜芦醇激活SIRT1可抑制神经祖细胞的增殖,引导其朝向星形胶质细胞系(而非神经元细胞系)分化,从而模拟了氧化状态的效应[269];而通过siRNA抑制SIRT1mRNA则可阻断白藜芦醇的效应。SIRT1的作用机制可能为,与发状分裂相关增强子1(Hes1)结合以去乙酰化H3K9,从而稳定转导蛋白样增强子蛋白1(TLE1)的辅阻遏因子复合体,而后者可抑制前神经元转录因子(proneuronal transcription factor)Mash1。另一项研究认为SIRT1可抑制Notch1-Hes1信号通路,SIRT1短暂转位到细胞核可能对神经祖细胞分化起一定作用[270]。因此,SIRT1可能从氧化应激、炎症、代谢等方面与神经祖细胞的分化发生联系。按照同样思路,Longo和同事报告[98],SIRT1缺失小鼠被氧化的蛋白和脂质水平降低,而SIRT1基因抑制小鼠则可对抗氧化应激。但对于增强SIRT1表达是否会有相反的效应则尚未明确。
SIRT1作为NF-κB的调节因子
近年来已将sirtuin作为免疫系统的新型调节因子,许多研究显示SIRT1可抑制多种组织的炎症程度[93,121,235,238,276,277]。NF-κB系统是适应性免疫和先天性免疫的主调节因子之一,该系统由受体和信号分子组成,且其功能在进化过程中高度保守。NF-κB为多效性转录因子,并作为危险信号(包括缺氧、氧化应激和遗传毒性应激)的胞浆感受器。NF-κB可与其他蛋白形成复合体,其中包括Rel蛋白家族成员(RelA/p65、c-Rel和RelB)以及NF-κB成分p50和p52。在基础状态,NF-κB复合体在胞浆可与NF-κB抑制因子(IκB)家族成员结合。而在激活状态,IκB被磷酸化,并成为泛素化的靶点而被降解,随后游离的NF-κB复合体转位至细胞核,触发靶基因(多数为促炎症因子)的表达。NF-κB结合域上调与增龄密切相关,而CR则可削弱NF-κB的活性[273]。SIRT1近来被认为是NF-κB系统的潜在抑制因子,从而为炎症与衰老提供了机制上的联系[275]。在转位至细胞核后,NF-κB的p65亚基被p300磷酸化,从而加强与DNA的结合。而SIRT1则可结合并去乙酰化RelA/p65,以抑制NF-κB的转录活性[202]。
sirtuin还可通过调节其他因子(如FOXO蛋白)以激活NF-κB信号;因为FOXO蛋白功能集中体现在胰岛素信号转导和应激通路,并可抑制NF-κB信号。过表达FOXO3a可抑制TNFα诱导的NF-κB激活,FOXO3a还可通过抑制NF-κB活性以促进凋亡[283]。有趣的是,多项研究报告发现,在线虫和哺乳动物系统,SIRT1可与FOXO蛋白相互作用并使之去乙酰化[118,187,285],提示NF-κB信号的调节可能有多个层面。
代谢紊乱与慢性炎症状态有关,可见脂肪组织和巨噬细胞释放促炎症细胞因子TNFα、IL-6,后两者可损害胰岛素在外周组织的作用。多项动物研究表明,SIRT1对代谢性疾病的有益功效,部分源自其可抑制脂肪组织和巨噬细胞的炎症。例如,过表达SIRT1和给予白藜芦醇的小鼠,其由高脂饮食所致的肝细胞炎症程度降低[93,95]。这些小鼠的NF-κB活性同样降低,与之相关的TNFα和IL-6等促炎症细胞因子的表达也减少[252]。调节SIRT1活性还可影响脂质在脂肪细胞的聚集,而脂质聚集则对肥胖和T2DM等代谢性疾病的发病造成不良影响。与上述情况相一致,肝脏特异性SIRT1缺失的小鼠经喂饲高脂饮食,其炎症表现和NF-κB信号转导增强[134]。与之相比,近期有研究采用全身SIRT1缺失小鼠发现,在其肾脏和肝脏有免疫球蛋白复合体的聚集,但未见免疫反应的变化[278]。
SIRT1与RelA/p65的相互作用可因吸烟而削弱,表现为巨噬细胞中NF-κB乙酰化加重并被激活,引发促炎症反应[279]。如上所述,给予白藜芦醇可减轻香烟烟雾提取物对大鼠动脉和CAEC的毒性效应,而SIRT1基因抑制后,白藜芦醇的此效应被阻止[238]。有趣的是,培养于正常饮食小鼠血清中的CAEC,可见NF-κB被激活和炎症基因表达。与之相比,给予CR小鼠血清的CAEC则显示ROS的生成、NF-κB的激活以及炎症基因的诱导均显著减少;而通过siRNA抑制SIRT1,则上述变化又会被削弱。因此,CR的血管抗氧化和抗炎症作用,可能是通过SIRT1依赖性机制,经循环因子的介导而实现[272]。
按照类似的思路,在吸烟者和慢性阻塞性肺疾病(COPD)患者肺部可见细胞核SIRT1水平较非吸烟者降低[280,281]。针对COPD烟雾小鼠模型,采用SRT2172(为选择性STAC)鼻内治疗,可阻止肺内中性粒细胞增多和运动耐量降低,明显抑制基质金属蛋白酶9(MMP-9)的增加。这项研究提示,激活SIRT1可能作为COPD等慢性炎症性疾病的有效治疗手段。
葡萄膜炎(Uveitis)为眼球中间层的炎症,在美国约占失明病例的10%。针对内毒素诱导性葡萄膜炎小鼠研究发现,白藜芦醇预处理可抑制眼部炎症区视网膜血管的白细胞粘附现象,且该抑制效应显著并呈剂量依赖性;还可见单核细胞趋化蛋白-1(MCP-1)、ICAM-1、8-羟基脱氧鸟苷(8-OHdG)和NF-κB水平显著降低,而视网膜色素上皮-脉络膜的细胞SIRT1活性则增加[282]。
有证据显示,人类免疫缺陷病毒-1(HIV-1)感染过程中可抑制SIRT1。过度活跃的免疫反应可增强HIV-1感染性和复制力,而研究提示NF-κB信号转导与之相关[286]。已知HIV的转录激活需要HIV-1反式转录激活因子(Tat)的参与,而在多个层面上,SIRT1均与这一途径相联系。首先,Tat本身即为SIRT1的靶点[287,288];两者可形成复合体,协同激活HIV启动子。SIRT1可将Tat去乙酰化以恢复其非乙酰化形态,使其与反式激活反应RNA和转录延伸因子相结合[288]。其次,Tat似乎还可调节SIRT活性。Tat可与SIRT1直接结合,阻断其去乙酰化p65的作用[288]。这种情况下,在野生型细胞中Tat可过度激活NF-κB的靶基因表达,而在SIRT1缺失细胞则未见。由此SIRT1、Tat和NF-κB形成闭合环路发挥作用,即SIRT1增强Tat的作用,而Tat则抵消SIRT1针对NF-κB信号的副作用。
SIRT6与免疫反应
调节免疫反应的sirtuin还有SIRT6,其可能为促长寿蛋白。SIRT6缺失小鼠存在严重的发育缺陷,表现为低血糖症和早衰表型[224]。近期发现,SIRT6可与NF-κB复合体的RelA/p65成分相互作用,还可被募集到NF-κB靶基因的某些启动区[289];在该区域,SIRT6去乙酰化组蛋白H3赖氨酸9,从而抑制NF-κB靶基因的表达。与之相一致,SIRT6缺失小鼠可见NF-κB的多个靶基因表达增强。另一独立研究发现,SIRT6可调节TNF的生成[290]。引人注目的是,SIRT6缺失小鼠的短寿和退行性表型可能源于NF-κB的过度激活,因为RelA单倍不足(haploinsufficiency)可代偿SIRT6敲除动物的早衰表型。
发现酵母SIR2能够延长寿命至今仅有10年时间。之后认识到SIR2只是一个高度保守的酶类大家族的典型成员,该家族可调节关键的细胞过程。例如,营养不足环境下能源物质的适应性调节,以及线粒体的生物合成和功能发挥,尤其与DNA修复、神经元存活和基因表达年轻态的保持有关。
然而,关于sirtuin的生物学研究仍有诸多问题有待揭示。例如,复杂的小鼠实验方案造成了研究数据的差异,尽管SIRT1的众多底物已被发现,但其相关功能还存在许多未知。困惑之处部分表现为,有关激活的SIRT1的研究数据在解释时存在相互冲突的现象。例如,肝脏SIRT1是通过禁食还是通过CR被诱导?现已明确SIRT1及其调节因子Nampt是呈12h周期变动的生物钟的关键成分,而越早的SIRT1研究是否越难以对此做出解释[51,53,291,292]?NAD+是sirtuin重要的生理性调节因子,甚至线粒体的NAD+也是如此[44],然而至今仍不清楚NAD+在不同的细胞区室中是如何合成的。还有,NAD+是否由NAM或NR生成,NAD+是否可作为NMN经细胞膜运输?
尽管sirtuin生物学研究突飞猛进,但尚无研究工具能够精确测定SIRT1体外对自然底物的活性,以及评价sirtuin的体内活性。目前研究者将sirtuin蛋白水平、NAD+浓度和sirtuin底物体内的乙酰化状态作为观察指标。通过将上述方法联合应用,即可获得大体的初步印象。然而由于sirtuin各靶点相互重叠(如SIRT1-3与FOXO蛋白),所以获得的情况并不完全。sirtuin活性还受到转译后修饰和抑制性蛋白(如DBC1对SIRT1)的调节[27,62]。因此,上述考虑事项凸显了评价手段的必要性,以便能够量化SIRT1-7的比活性(specific activity)。
最为重要的问题是,激活SIRT1是否足够安全以用于人类疾病(如糖尿病、CVD)的治疗。很明显,CR存在某些弊端(如生殖能力降低、骨质疏松),然而对于过表达SIRT1小鼠或给予STAC的小鼠或人类,并未报告有这些副作用[94,96,114,293]。既然STAC针对多种疾病已开展临床试验,我们应该能在未来数年内获取上述问题的答案。
与10年前最初开展哺乳动物sirtuin研究时相比,目前我们面临着巨大挑战。能量摄入与机体健康和存活之间存在着精细的协调机制,而sirtuin研究无疑将会大大丰富我们对这一机制的认识。在未来十年,我们应能够明确sirtuin是否可作为治疗增龄性疾病的安全靶点。
公开声明(disclosure statement)
David A. Sinclair为Srtris公司顾问,该公司是葛兰素史克(GSK)子公司,目标为开发以sirtuin为靶点治疗增龄性疾病的药物。
志谢(acknowledgments)
Marcia C. Haigis获得衰老科研基金布鲁克代尔领导项目(Brookdale Leadership in Aging Fellowship)和保罗·F·格伦(Paul F. Glenn)基金会资助。DavidA. Sinclair获得美国国立卫生研究院(NIH)、PaulF. Glenn基金会和埃利森(Ellison)医学基金会资助。
哺乳动物去乙酰化酶sirtuin的生物学特征及疾病相关性(Mammalian sirtuins:biologica.pdf
https://blog.sciencenet.cn/blog-426290-900309.html
上一篇:衰老的特征 The Hallmarks of Aging
下一篇:衰老遗传学(The genetics of ageing)