博文
释氢纳米酶自维持催化免疫治疗
||
释氢纳米酶自维持催化免疫治疗
释氢纳米酶构建线粒体活性氧放大器用于自维持催化免疫治疗
本研究立足于氢气不仅能通过选择性抗氧化保护细胞和线粒体,又不影响安全活性氧水平,有利于维持安全的氧化应激水平。类似于消防员穿着的防护服,避免自身损伤,把灭火器活性氧对付肿瘤细胞。利用材料学技术,通过催化产氢气的材料保护线粒体以产生更多活性氧的策略实现自催化免疫治疗。
纳米催化疗法是肿瘤精准医学领域的关键研究方向[1−4]。该疗法利用纳米材料模拟酶催化反应,在肿瘤微环境(TME)中原位产生具有细胞毒性的活性氧(ROS),从而实现对癌细胞的选择性杀伤。与传统治疗手段相比,该策略可由特定肿瘤微环境条件(如弱酸性、过氧化氢(H₂O₂)水平高于正常组织)激活,具备特异性高、全身毒性低、耐药风险小等显著优势[5−9]。然而,该方法的临床转化受限于一系列核心难题,包括肿瘤内催化底物不足、抗氧化防御系统活性过高等。因此,单一催化过程产生的活性氧强度弱、持续时间短,难以引发肿瘤细胞大规模死亡。此外,肿瘤微环境的异质性与免疫抑制特性,也会削弱催化治疗的彻底性与全身疗效[10−14]。
值得注意的是,线粒体作为细胞能量代谢的核心枢纽与活性氧的主要来源,对催化治疗效果起决定性作用。传统策略中,剧烈且短暂的外源性活性氧爆发常导致线粒体膜电位快速崩溃、电子传递链功能紊乱,伴随ATP耗竭并迅速启动细胞凋亡[15−18]。尽管这种急性损伤可诱导部分细胞死亡,但同时会使线粒体丧失内源性活性氧生成能力,限制治疗的有效性与持久性。因此,将治疗策略从“破坏线粒体”转向“重编程线粒体功能,使其成为持续产生内源性活性氧的发生器”,是提升纳米催化疗法疗效的重要方向。
近年来,氢气(H₂)疗法备受关注。氢气具有生物安全性高、细胞膜穿透性优异及选择性抗氧化等特性[19−23],可轻易穿透线粒体等亚细胞结构。更重要的是,氢气能够缓解氧化应激所致的线粒体损伤,维持膜相对稳定,延缓电子传递链崩溃,并调控线粒体代谢与能量平衡。通过将线粒体功能从“急性衰竭”重编程为“亚致死性损伤”状态,氢气既可保护正常组织,又能诱导活性氧持续泄漏[24,25],显著延长并放大肿瘤治疗中内源性活性氧的生成。氢气疗法与纳米催化联用可构建自增强的治疗体系:催化反应持续生成杀伤肿瘤的活性氧,氢气则通过选择性抗氧化调控活性氧分布与线粒体功能,二者协同实现更彻底、更持久的治疗效果。
目前,已有多种释氢纳米材料应用于肿瘤治疗,但现有体系仍存在明显缺陷。金属氢化物(如CaH₂、MgH₂)[26]储氢量高,但化学稳定性差,在生理循环中易发生非特异性氢气泄漏;无机非金属载体材料(如聚多巴胺基体系)[27]稳定性与载氢量有所提升,但其释氢多依赖内源性或外源性催化,存在组织穿透深度有限、调控精度不足等问题;氢化钯(PdH)等经典金属储氢材料虽具治疗潜力[28],但难以在单一体系中同时实现合成简便、高储氢量、高稳定性与按需可控释氢;而CdS、TiO₂等光解水半导体材料[29]则普遍面临光腐蚀严重、生物相容性差、产氢效率低等难题。
本研究开发的掺氢铑钯合金(RhPd‑H)纳米酶可有效解决上述局限:(1)储氢稳定性优异,在常温常压下无明显释放,克服了传统金属氢化物不稳定的问题;(2)具备独特的近红外(NIR)光响应特性,可实现时空可控的按需释氢,精准度远超非金属载体体系;(3)与其他通常兼具过氧化氢酶(CAT)样、过氧化物酶(POD)样活性或广谱抗氧化功能的钯氢基纳米酶不同[23,30,31],RhPd‑H通过铑掺杂实现高选择性单一过氧化物酶活性,避免了过氧化氢的无效消耗,可将内源性H₂O₂高效转化为毒性羟基自由基(·OH),构建清洁高效的促氧化治疗机制。
综上,RhPd‑H可将氢气疗法、光热疗法与纳米催化疗法整合于单一平台。在近红外光单一刺激下,光热疗法产生的局部热效应既可提升催化反应速率,又能触发氢气快速释放;同时,催化反应消耗H₂O₂生成·OH,引发初始氧化爆发。释放的H₂扩散至线粒体并清除活性氧,已有研究证实释氢纳米材料可有效降低肿瘤细胞内活性氧水平[27,32]。值得注意的是,该清除作用具有瞬时性,初始活性氧降低后,持续氢气暴露可重编程线粒体功能,诱导内源性活性氧持续生成[33]。这种“先保护、后功能重编程”的双相调控模式,在部分保留线粒体结构的同时造成电子传递链效率下降,形成“泄漏”状态以维持长期氧化应激。由此,线粒体被重编程为内源性活性氧“次级放大器”,与催化产生的外源性活性氧形成正反馈循环,最终引发强效持久的活性氧爆发,高效诱导细胞凋亡(示意图1)。
该体系实现了时空同步的“催化-光热-氢气”三重协同策略,显著增强肿瘤细胞杀伤效果,并通过免疫原性细胞死亡(ICD)、树突状细胞(DCs)成熟与T细胞活化启动强效抗肿瘤免疫循环。本研究揭示了氢气对线粒体功能的精准调控是增强纳米催化免疫治疗的新机制,为多模式催化治疗提供了全新范式。
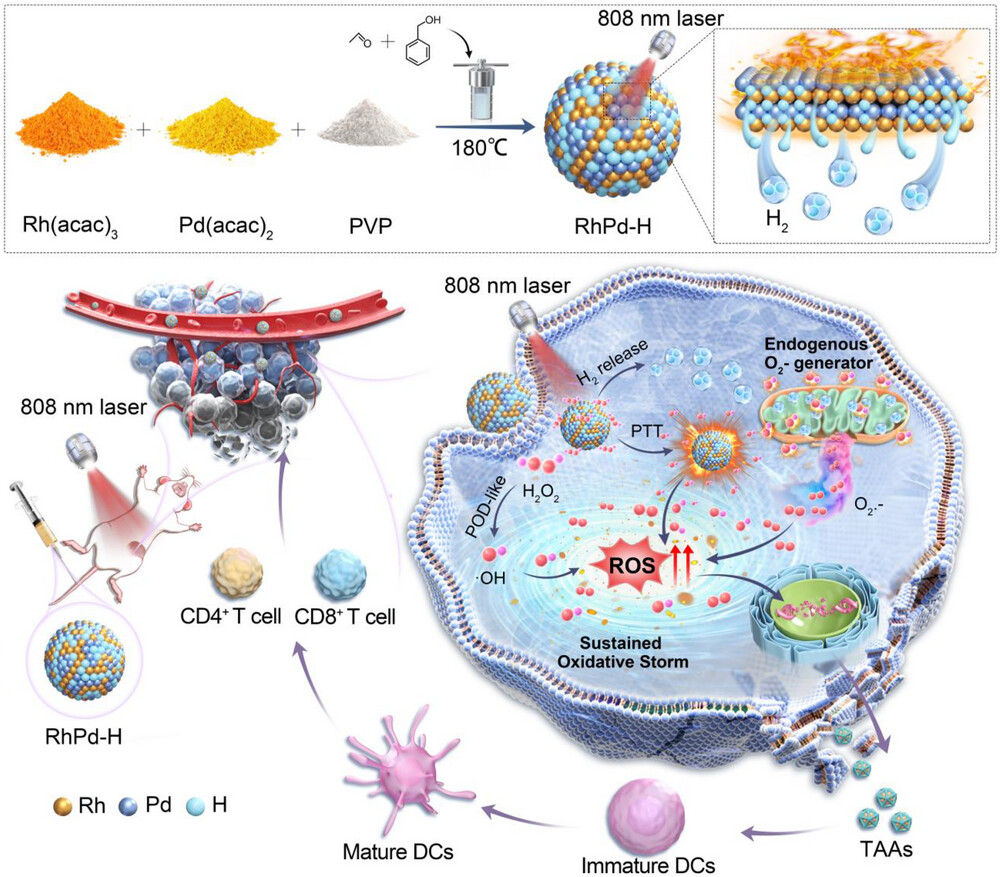
示意图1
RhPd‑H纳米酶调控的可编程级联反应:自放大氧化风暴与抗肿瘤免疫
近红外光照激活RhPd‑H纳米酶,同步发挥光热作用与过氧化物酶模拟催化功能,引发初级氧化应激;热触发释放的氢气靶向线粒体,将其重编程为持久的内源性活性氧放大器,驱动级联式、自维持的氧化风暴。这种持续氧化应激可诱导强烈的免疫原性细胞死亡,促进肿瘤相关抗原(TAAs)与损伤相关分子模式(DAMPs)释放,激活树突状细胞并招募细胞毒性T淋巴细胞,将局部纳米催化治疗转化为全身抗肿瘤免疫应答。
Shi M, Cao J, Wu T, Yang G, Yang Y, Zhang S, Su W, Chu H, Zhao Y, Jiang S, Wu Q, Jiao D, Chen F. A Hydrogen-Releasing Nanozyme Engineers a Mitochondrial ROS Amplifier for Self-Sustaining Catalytic Immunotherapy. Adv Sci (Weinh). 2026 Apr 10:e24313.
2 结果与讨论
2.1 RhPd‑H 纳米颗粒的表征
本研究对合成的 RhPd‑H 纳米结构进行了系统表征,以确认其基本结构、元素组成、稳定性及生物相容性。X 射线光电子能谱(XPS)分析证实成功制备了高纯度 Rh‑Pd 复合物,未检测到杂质峰(图 1A)。透射电子显微镜(TEM)观察显示,RhPd‑H 呈不规则多面体形貌,平均粒径为 16 ± 2 nm(图 1B)。高角环形暗场扫描透射电镜(HAADF‑STEM)元素 mapping 结果表明,单个纳米颗粒内 Rh 与 Pd 信号分布高度均匀且显著重叠(图 1C),呈现清晰的共定位特征,证实成功形成 Rh‑Pd 合金结构,而非核壳结构或物理混合物。
Zeta 电位测试表明,铑钯合金(RhPd)与 RhPd‑H 在去离子水中均带负电,电位分别约为 −5.35 mV 与 −15.36 mV(图 S1)。RhPd‑H 显著更高的电负性可能源于氢化作用对表面电子结构的调控,有利于阴离子或羟基吸附,从而提升其在水相环境中的胶体稳定性。我们进一步评估了 RhPd‑H 的胶体行为。如图 S2 所示,RhPd‑H 纳米颗粒在水、PBS 及含 10% 胎牛血清的 1640 培养基中均保持良好稳定性,无明显聚集,为后续生物学评价提供了可靠基础。
通过溶血实验评估了 RhPd‑H 的生物相容性。将新鲜红细胞与不同浓度的 RhPd‑H 纳米颗粒共孵育,在 540 nm 处测定上清液吸光度。结果显示,即使浓度达到 60 µg/mL,溶血率仍低于 3%(图 S3),为该材料的静脉注射与体内应用提供了重要的安全性依据。
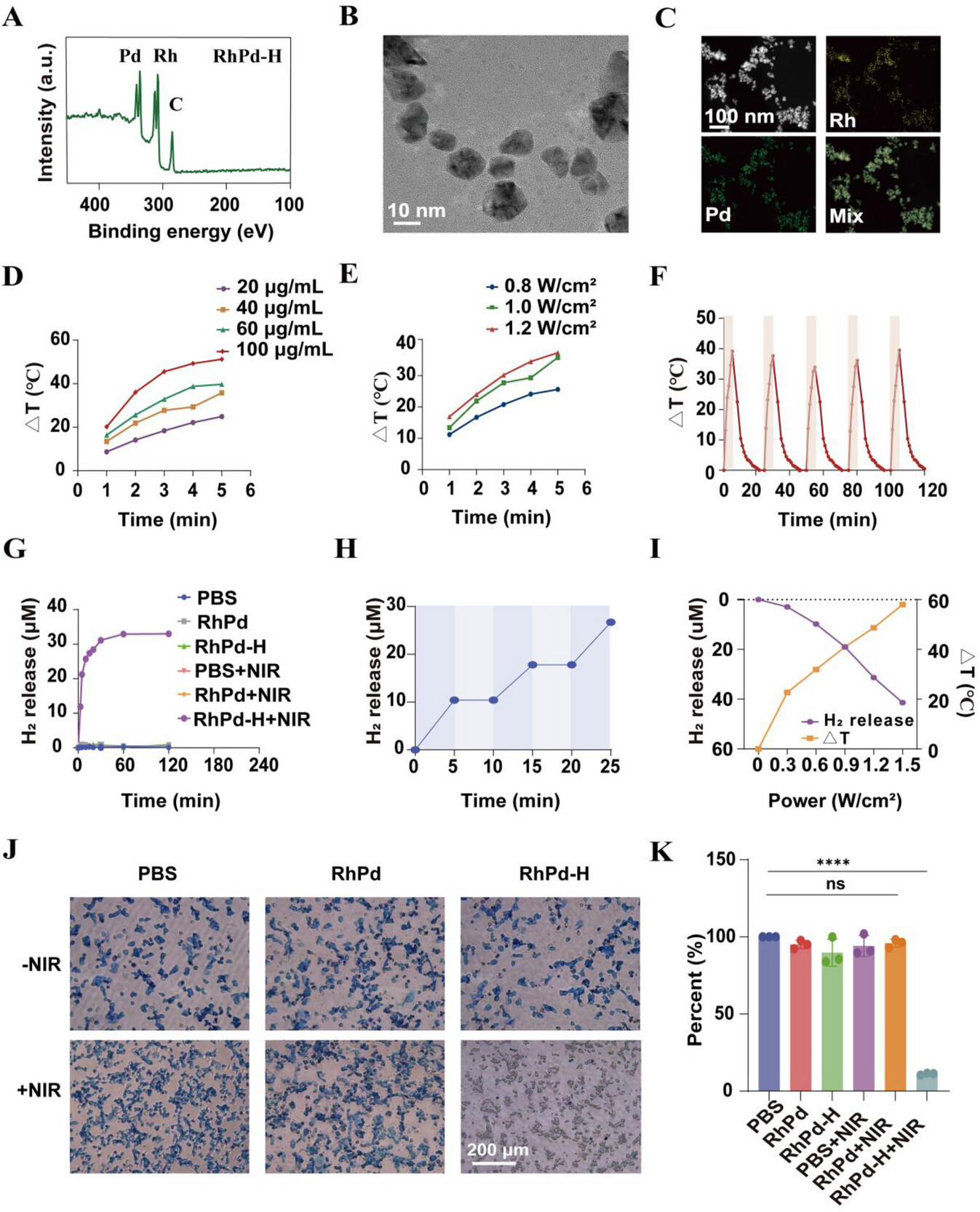
图 1 RhPd‑H 纳米酶的表征与性能测试
(A) RhPd‑H 纳米颗粒的 XPS 图谱;(B) TEM 图像;(C) HAADF‑STEM 与 EDX 元素 mapping 图像。
(D) 808 nm 激光(1 W/cm²)照射下,PBS 中不同浓度 RhPd‑H 的升温曲线;
(E) 不同功率密度 808 nm 激光照射下 RhPd‑H 的升温曲线;
(F) RhPd‑H 的光热循环测试;
(G) 不同处理条件下的氢气释放行为;
(H) 激光开关控制下 RhPd‑H 氢气释放的动力学与可逆性;
(I) 不同功率下 RhPd‑H 的释氢量与温度变化曲线;
(J) 亚甲基蓝(MB)探针法可视化 4T1 细胞内产氢;
(K) MB 法定量细胞内 H₂ 水平(n = 3)。
采用双侧 Student’s t 检验进行统计学分析:*p < 0.05,**p < 0.01,***p < 0.001,****p < 0.0001。
---
2.2 光热触发氢气释放性能评价
RhPd‑H 纳米颗粒的光热升温行为呈现显著的浓度依赖性与功率依赖性(图 1D、E)。在近红外激光(808 nm,1 W/cm²)照射下,当浓度从 20 提升至 100 µg/mL 时,溶液平衡温升从 24.9 ℃ 升至 51.2 ℃。类似地,在固定浓度(60 µg/mL)下,温升随激光功率(0.8–1.2 W/cm²)呈线性增长,最大温升 ΔT ≈ 37.3 ℃。
在中等浓度 40 µg/mL、相同照射条件(1 W/cm²,5 min)下的补充实验中,RhPd 表现出相似趋势,证实其可作为高效光热转换核心(图 S4)。此外,该材料表现出优异的光热稳定性。在连续 4 次“激光开启 10 min‑激光关闭自然冷却 10 min”循环中,RhPd‑H 每次循环的最大温差均小于 5.6 ℃,升温/降温曲线高度可重复(图 1F),表明材料经多次光热循环后微观结构与光学性质基本保持不变,具备良好光热稳定性。
基于 10 min 内的升降温曲线,计算得到 RhPd‑H 的光热转换效率高达 η ≈ 65.18%(图 S5),优于多数已报道的贵金属基光热材料。这种高能量转换能力主要源于 RhPd 合金纳米结构的局域表面等离子体共振(LSPR)效应及其在第一近红外窗口的宽谱吸收,凸显其在光热治疗应用中的巨大潜力。
光学显微镜观察显示,近红外照射下 RhPd‑H 水溶液中产生大量微气泡,而相同条件下 RhPd 纳米颗粒无气泡产生(图 S6)。此外,亚甲基蓝还原实验证实,仅 RhPd‑H 在激光照射下产生还原性 H₂ 气体,使染料高效褪色。定量结果显示,照射 60 min 后 664 nm 处吸光度下降 50.61%,对应累积释氢量 32.9 µM;相比之下,RhPd 纳米颗粒几乎不引起 MB 还原(图 1G)。
这些对照实验有效排除了光催化水分解作为主要贡献途径,表明 RhPd‑H 的近红外触发释氢主要由氢化物晶格的光热分解驱动。为进一步探究释放机制,我们通过改变激光功率研究温升与析氢的相关性。溶液温度与产氢量随激光功率同步升高,直接证实光热效应是释氢的关键驱动力(图 1I)。即光热转换产生的局域加热破坏 RhPd‑H 中的金属‑氢键,促进晶格氢脱附并复合为 H₂ 分子。
此外,RhPd‑H 的近红外触发释氢在激光反复开关循环下表现出高度可控性。采用 808 nm 激光交替照射(5 min 开/5 min 关),仅在照射阶段发生可重复产氢,激光关闭后产氢立即停止。这种可重复的开关行为证明 RhPd‑H 具备精准可靠的光响应可控释氢性能(图 1H)。
在细胞水平,将经 MB 预染色的 4T1 细胞用 RhPd‑H 处理并照射 5 min,60 min 内观察到明显的胞内褪色现象,荧光强度下降约 90%,表明光热触发释氢及氢气介导的胞内还原作用有效发生(图 1J、K);而各对照组均无明显荧光降低。综上,RhPd‑H 可在近红外光下通过 RhPd 核心光热转换与氢化钯热分解机制实现可控、具有生物还原活性的氢气释放,为基于氢气的生物学调控提供了有力工具。
---
2.3 氢气增强本征过氧化物酶(POD)模拟催化机制
在 pH 6.5 酸性条件下,RhPd‑H 纳米颗粒表现出显著的 POD 模拟活性(图 2A)。其可催化 H₂O₂ 分解产生·OH,氧化底物 TMB 形成蓝色氧化产物(oxTMB),在 625 nm 处出现特征吸收峰,且呈浓度依赖性(图 2B)。催化活性具有强烈的 pH 依赖性,在 pH 5 时效率最高,pH 6.5 与 7.4 时逐步下降(图 2C)。这种 pH 响应行为与天然过氧化物酶偏好酸性微环境的特性一致,表明 RhPd‑H 具备在酸性肿瘤区域实现靶向催化的潜力。
在 pH 6.5 条件下,通过改变 H₂O₂ 与 TMB 浓度对 RhPd‑H 纳米酶进行动力学分析,采用米氏模型与双倒数图计算相应动力学参数。以 TMB 为底物时,米氏常数(Km)为 1.034 mM,最大反应速率(Vmax)为 7.63×10⁻⁹ M·s⁻¹(图 2E);以 H₂O₂ 为底物时,Km 与 Vmax 分别为 6.911 mM 与 8.27×10⁻⁹ M·s⁻¹(图 2F)。电子顺磁共振(EPR)光谱进一步证实·OH 生成,RhPd‑H 与 RhPd 体系均出现典型的 DMPO‑·OH 信号(图 2D)。
紫外‑可见吸收光谱与 EPR 表征结果表明,在同等质量浓度下,RhPd‑H 的 POD 模拟活性优于 RhPd。此外,在一系列 H₂O₂ 浓度下的测试一致显示,所有条件下 RhPd‑H 的类 POD 活性均高于 RhPd(图 2G)。上述结果表明,位于金属晶格中的氢原子可有效增强类 POD 催化活性。
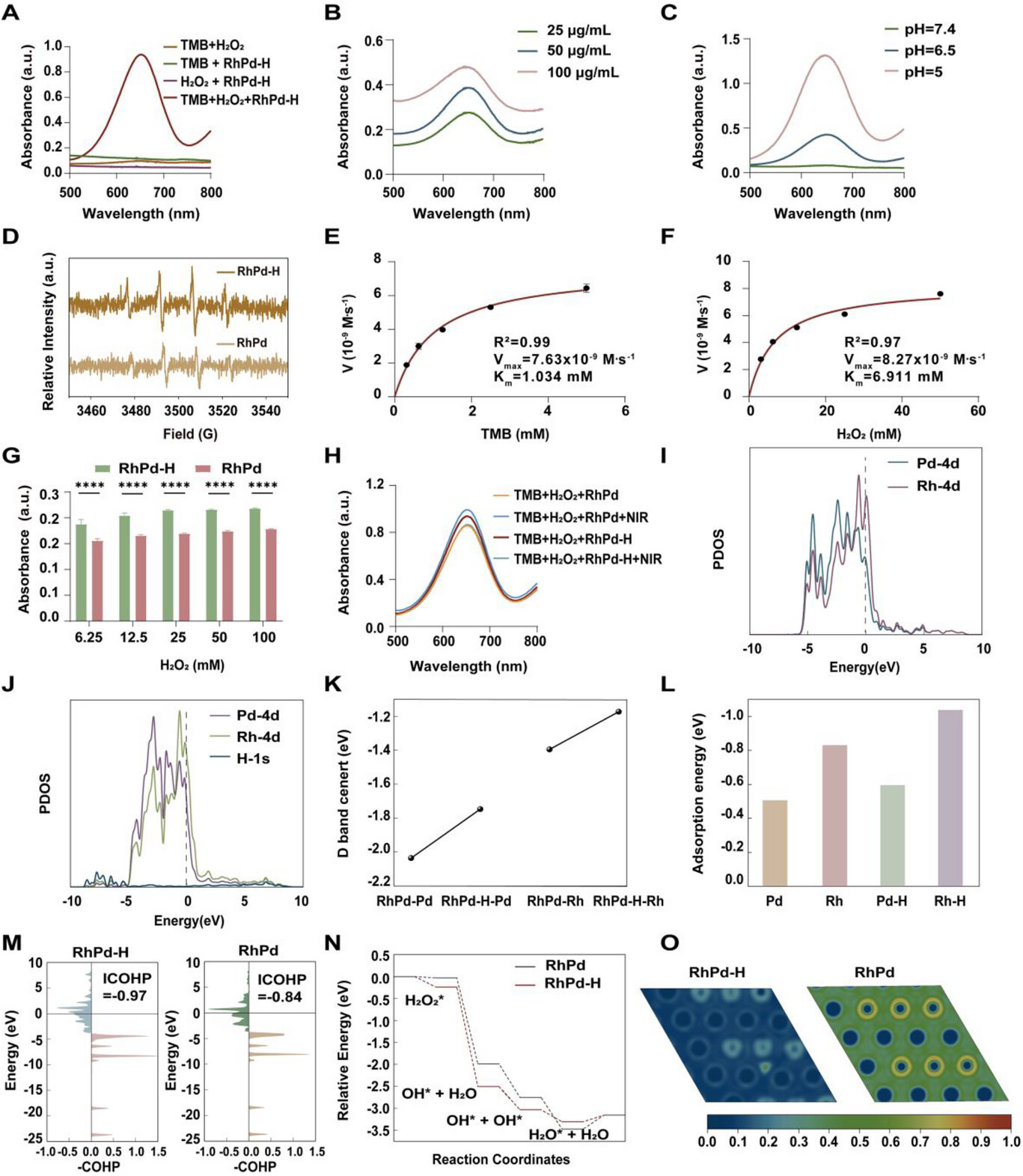
图 2 氢气增强类 POD 活性与 DFT 模拟
(A–C) RhPd‑H 类 POD 活性的浓度与 pH 依赖性;
(D) pH 6.5 缓冲液中加入 H₂O₂ 后 RhPd‑H 与 RhPd 纳米颗粒的 ESR 图谱;
以 (E) TMB、(F) H₂O₂ 为底物的 RhPd‑H 纳米酶米氏动力学曲线;
(G) 不同 H₂O₂ 浓度下 RhPd‑H 与 RhPd 类 POD 活性对比(n = 3);
(H) 有无近红外照射下 RhPd‑H 与 RhPd 类 POD 活性对比;
RhPd (I) 与 RhPd‑H (J) 纳米颗粒的态密度(PDOS);
(K) Pd 与 Rh 位点的 d 带中心;
(L) RhPd 与 RhPd‑H 纳米颗粒对 H₂O₂ 的吸附能;
(M) RhPd‑H 与 RhPd 纳米颗粒的晶体轨道哈密顿布居(COHP);
(N) RhPd‑H 与 RhPd 纳米颗粒类 POD 反应的自由能图;
(O) RhPd‑H 与 RhPd 纳米颗粒表面的电子定域化函数(ELF)。
统计学分析采用双侧 Student’s t 检验:*p < 0.05,**p < 0.01,***p < 0.001,****p < 0.0001。
近红外照射下两种材料行为出现差异:RhPd 的类 POD 活性因光热加速反应动力学而提升,oxTMB 吸光度上升;而 RhPd‑H 释放的氢气会消耗部分原位生成的·OH,部分抵消光热加热带来的加速效应,导致 oxTMB 信号略有下降(图 2H)。
为进一步探究氢气增强本征类 POD 催化的内在机制,进行了密度泛函理论(DFT)计算。通过对比投影态密度(PDOS)阐明 RhPd 与 RhPd‑H 催化剂的电子结构。如图 2I 所示,RhPd 中 Pd 与 Rh 的 d 带呈现宽而连续的峰分布;H‑1s 轨道与 Pd/Rh 4d 轨道发生杂化,证实 RhPd‑H 中形成稳定化学键(图 2J)。同时,氢掺杂使金属 d 带变锐并抬高铁米能级(Ef),提升电化学活性,显著改善 RhPd 金属烯的类过氧化物酶性能。
此外,氢掺杂使 Pd 与 Rh 的 d 带中心均向上移动:Pd 从 −2.04 eV 移至 −1.75 eV,Rh 从 −1.407 eV 移至 −1.1 eV(图 2K),增强电活性并强化 H₂O₂ 吸附。吸附能计算(图 2L)证实这一趋势:RhPd‑H 对 H₂O₂ 的吸附能更负,结合更强;两种金属位点中 Rh 对 H₂O₂ 亲和力更高,是催化过程中捕获 H₂O₂ 的主要活性中心。
为验证 Rh 位点与 H₂O₂ 的结合强度,计算晶体轨道哈密顿布居(COHP,图 2M)。RhPd‑H 与 H₂O₂ 的相互作用显著强于 RhPd,成键态更优;其积分 COHP(ICOHP)值为 −0.97 eV,进一步支持该结论。基于类 POD 反应机理,计算关键基元步骤的自由能变化(图 2N),结果表明 RhPd‑H 在 H₂O₂ 还原过程中热力学更有利;类 POD 反应中的 H₂O 脱附步骤对 RhPd‑H 更友好,仅需 0.28 V,而 RhPd 需 0.46 V。
此外,RhPd‑H 与 RhPd 纳米颗粒表面的电子定域化函数(ELF)分析显示,氢掺杂导致表面金属原子电荷显著降低(图 2O),使 RhPd‑H 向 H₂O 转移的电子更少,削弱 H₂O 吸附,从而促进后续催化循环。综上,将氢原子引入 RhPd 金属烯可调控其电子性质、促进 H₂O₂ 吸附、优化反应中间体结合、降低水脱附能垒,共同增强类 POD 催化活性。
鉴于 Pd‑H 材料通常同时具有过氧化氢酶(CAT)与超氧化物歧化酶(SOD)样活性,而本实验表明 RhPd‑H 不具备这两种活性,我们通过理论计算分析 Pd‑H 与 RhPd‑H 表面两条反应路径的自由能分布。超氧阴离子(·O₂⁻)作为布朗斯特碱易从水中夺取质子形成氢过氧自由基(OOH),因此选用 OOH 作为评估 SOD 样活性的起始反应物。如图 S7A 所示,Pd‑H 对 OOH 的结合弱于 RhPd‑H;在 SOD 样催化循环中,OOH 需进一步质子化或歧化,但 RhPd‑H 对 OOH 过强的结合使中间体稳定化,阻碍后续转化,导致 SOD 样活性丧失。
对于 CAT 样活性,H₂O₂ 与 O₂ 从 Pd‑H 表面脱附比从 RhPd‑H 更有利(图 S7B),表明产物脱附在 RhPd‑H 表面受阻,无法实现活性位点再生,因此不具备 CAT 样活性。上述结果表明,Rh 掺杂可选择性调控纳米酶活性:尽管 Pd‑H 本征具有 POD、CAT 与 SOD 三重活性,引入 Rh 后仅增强类 POD 活性并抑制另外两种。这种专一保留类 POD 活性的特性使 RhPd‑H 避免多酶活性竞争,实现更高催化效率。
---
2.4 氢气编程的胞内自维持氧化风暴
采用 CCK‑8 法系统评估了不同浓度与孵育时间下 RhPd‑H 对 4T1 与 MCF‑7 细胞的毒性。如图 3A 所示,RhPd‑H 对 4T1 细胞的毒性呈时间与浓度依赖性升高。40 µg/mL 浓度共孵育 24 h 后,4T1 细胞存活率降至 68%,表明 RhPd‑H 介导的纳米催化治疗可对该细胞系产生细胞毒性。相比之下,相同条件下 RhPd‑H 对 MCF‑7 细胞无明显毒性。
我们进一步在 808 nm 激光照射下评估其体外光毒性。激光照射后,RhPd‑H 对 4T1 与 MCF‑7 细胞均表现出显著的浓度依赖性细胞毒性;当浓度升至 40 µg/mL 时,4T1 与 MCF‑7 细胞存活率分别降至 9.72% 与 58.99%(图 3A、B)。
为进一步探究氢气在提升材料抗癌效果中的作用,对比了不同浓度下 RhPd 与 RhPd‑H 在 808 nm 激光照射下对 4T1 与 MCF‑7 细胞的影响。如图 3C、D 所示,两种材料均对两种癌细胞产生浓度依赖性毒性;值得注意的是,在毒性浓度范围内,RhPd‑H 的抗肿瘤效果显著优于 RhPd 且呈剂量依赖性,高浓度下优势更明显。例如 40 µg/mL 时,RhPd‑H 组 4T1 细胞存活率仅为 RhPd 组的 16.31%。结果表明,氢掺杂显著增强光热与纳米催化的联合治疗效果,且在一定浓度阈值以上提升尤为明显。
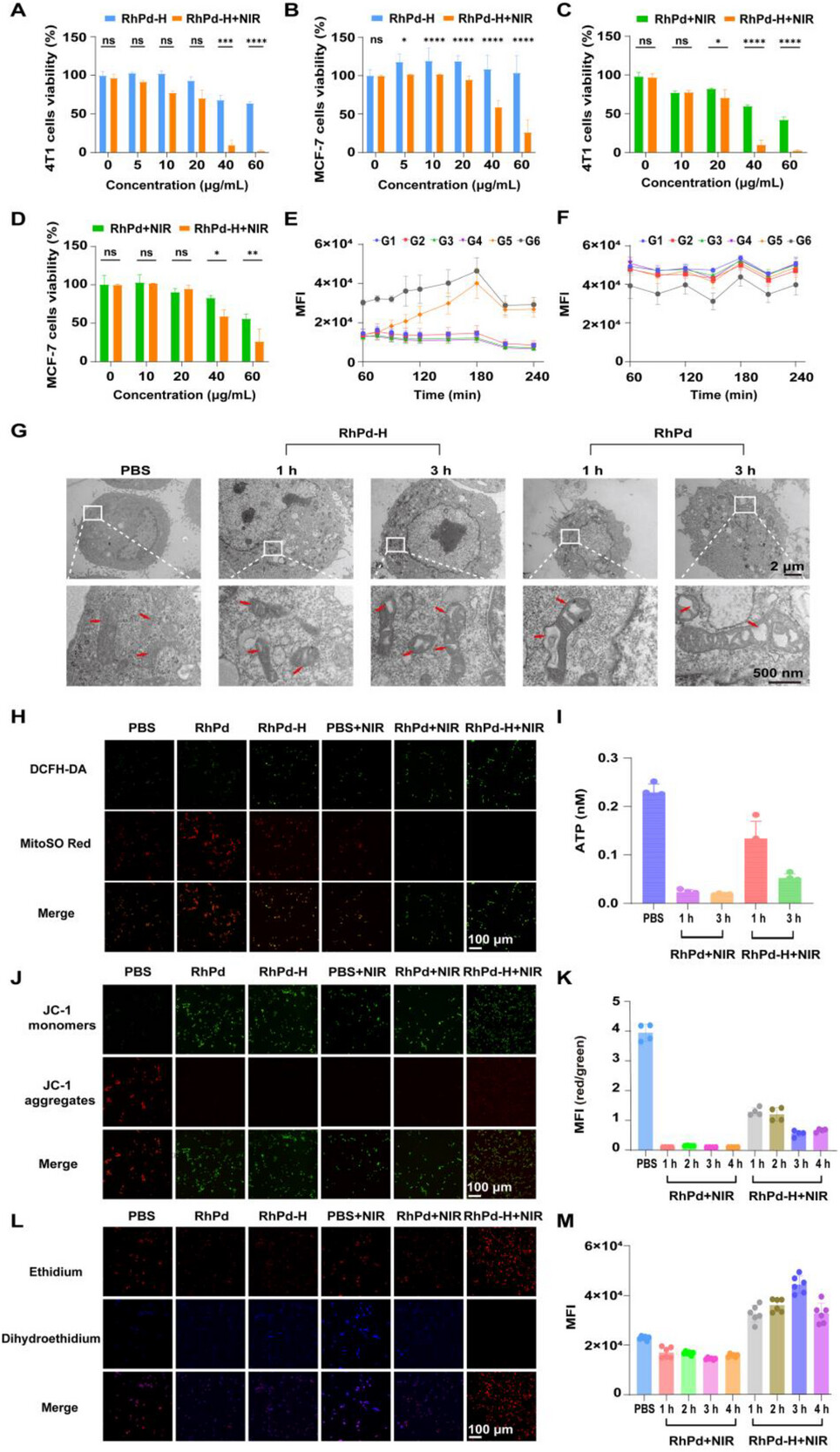
图 3 RhPd‑H 纳米酶诱导持续性氧化风暴的机制
(A–D) 不同浓度 RhPd 或 RhPd‑H 处理 (A、C) 4T1 与 (B、D) MCF‑7 细胞的存活率:(A、B) 无近红外照射,(C、D) 有近红外照射(n = 3);
不同处理下 (E) DCFH‑DA 与 (F) MitoSOX Red 平均荧光强度随时间的变化(n = 5);
处理组:G1,PBS;G2,RhPd;G3,RhPd‑H;G4,PBS+NIR;G5,RhPd+NIR;G6,RhPd‑H+NIR;
(G) 1 h 与 3 h 时 RhPd+NIR 与 RhPd‑H+NIR 处理后 4T1 细胞线粒体结构的 TEM 图像;
(H) 2 h 时不同处理(G1–G6)下 4T1 细胞的 DCFH‑DA/MitoSOX Red 双染结果;
(I) 1 h 与 3 h 时 RhPd+NIR 与 RhPd‑H+NIR 处理后 4T1 细胞的 ATP 水平检测(n = 3);
(J、L) 2 h 时不同处理下 4T1 细胞 (J) JC‑1 与 (L) DHE 染色的共聚焦激光扫描显微镜(CLSM)图像;
(K、M) 1、2、3、4 h 时 RhPd+NIR 与 RhPd‑H+NIR 处理下 (K) JC‑1 红/绿荧光比(n = 4)与 (M) DHE 平均荧光强度随时间的变化(n = 6)。
统计学分析采用双侧 Student’s t 检验:*p < 0.05,**p < 0.01,***p < 0.001,****p < 0.0001。
为探究近红外照射下 RhPd‑H 纳米颗粒的协同抗肿瘤机制,系统评估其对 4T1 肿瘤细胞内 ROS 水平与线粒体功能的影响。采用 DCFH‑DA 探针检测胞内总 ROS,结果显示 3 h 后,与 PBS 对照组相比,RhPd+NIR 组荧光强度提升约 2.77 倍,而 RhPd‑H+NIR 组进一步升至约 3.20 倍(图 3E),表明 RhPd‑H+NIR 可诱导更显著的胞质 ROS 累积。
采用线粒体超氧阴离子指示剂 MitoSOX Red 定量线粒体来源的主要 ROS(·O₂⁻)。RhPd‑H+NIR 组荧光信号较 RhPd+NIR 组降低约 14.16%(图 3F),提示线粒体氧化应激明显下降。4T1 细胞 DCFH‑DA/MitoSOX Red 双染实验进一步证实,RhPd‑H+NIR 可同时实现胞质 ROS 升高(DCFH‑DA 荧光提升约 8.49 倍)与线粒体 ROS 抑制(MitoSOX Red 信号下降约 89%),展现出空间差异化调控 ROS 的能力(图 3H)。
为深入解析支撑这种差异化 ROS 调控的线粒体功能状态,首先通过 TEM 观察线粒体超微结构完整性(图 3G)。PBS 对照组线粒体嵴清晰完整,而 RhPd+NIR 与 RhPd‑H+NIR 组线粒体均出现不同程度空泡化,表现为肿胀与结构损伤;这种形态损伤具有时间依赖性且受氢气调控。在 1 h 与 3 h 两个时间点,RhPd‑H+NIR 组空泡化程度均显著轻于不含氢的 RhPd+NIR 组;且随时间推移损伤加重,3 h 样品空泡化均比 1 h 更明显。该直观结果直接证明,氢气可在应激条件下不完全保留线粒体结构,为后续功能观察提供形态学基础。
随后评估这种结构受损状态下的功能变化。采用 JC‑1 探针检测线粒体膜电位(ΔΨm),动力学曲线差异显著:与 PBS 组相比,RhPd+NIR 处理 1 h 内即引发 ΔΨm 急剧崩溃,红/绿荧光比下降约 39 倍;而 RhPd‑H+NIR 组下降更平缓,虽 1 h 已显著降低,但在 3 h 与 4 h 仍持续逐步衰减。3 h 时,RhPd‑H+NIR 组荧光比为 RhPd+NIR 组的 7.1 倍(图 3K)。
处理 2 h 后的共聚焦成像提供直观视觉证据:RhPd‑H+NIR 组仍可见明显红色荧光,而 RhPd+NIR 组仅呈现绿色荧光(图 3J)。上述结果明确证实,氢气可延缓线粒体膜电位丧失。
胞内 ATP 水平检测进一步支持线粒体“功能受损但持续运转”的状态。1 h 时,RhPd‑H+NIR 组 ATP 水平显著低于 PBS 对照组(约为对照的 58%),但远高于 ATP 耗竭的 RhPd+NIR 组(约为其 5.8 倍),表明尽管线粒体无法高效产能,仍保持代谢活性并维持基础功能。至 3 h 时,RhPd‑H+NIR 组 ATP 水平显著降至对照的 22.7%,但仍为 RhPd+NIR 组的 2.7 倍,表明该受损状态最终失效,但较无氢处理组能量崩溃明显延迟(图 3I)。
尽管氢气对线粒体具有部分保护作用,却无法完全逆转电子传递链的内在功能障碍,最终导致细胞大量产生·O₂⁻。二氢乙啶(DHE)共聚焦成像显示,RhPd‑H+NIR 处理可触发内源性超氧阴离子大量生成,荧光强度约为 RhPd+NIR 组的 6.8 倍(图 3L、M),证实线粒体作为主要来源持续产生·O₂⁻。
基于上述实验结果,我们提出 RhPd‑H 纳米颗粒通过氢气介导线粒体功能重编程实现强效肿瘤细胞杀伤。近红外照射下,RhPd‑H 同步实现光热治疗、纳米催化与氢气释放:光热与纳米催化在胞质协同引发·OH 爆发;同时释放的氢气扩散至线粒体并清除 ROS,减轻线粒体氧化损伤、维持膜电位稳定。
与传统光热/催化策略快速诱导线粒体彻底破坏不同,氢气的保护作用使线粒体进入结构保留但功能受损的特殊状态,表现为中等水平 ATP 产生。电子传递链效率下降导致电子泄漏增加,引发超氧阴离子持续生成,使线粒体转化为持续性内源性 ROS 发生器。
值得注意的是,这种氢气介导的线粒体调控与另一促氧化通路协同作用:已有研究证实 H₂ 可在肿瘤微环境中被催化转化为一氧化碳(CO),启动快速促氧化信号。该 CO 介导机制与本文所述线粒体重编程并不矛盾,而是形成互补:CO 快速生成提供初始急性氧化触发,而氢气诱导的线粒体氧化还原动力学改变构建长期功能障碍状态。受损线粒体不发生即时崩溃,而是持续维持高代谢与氧化应激水平。
因此,CO 快速信号与线粒体持续功能障碍两条通路协同,形成更剧烈、更持久的氧化风暴,维持胞内总 ROS 高水平并显著延长氧化应激,最终超出细胞抗氧化修复能力,引发不可逆脂质过氧化与细胞死亡。这种从“急性氧化休克”到“持续性氧化风暴”的策略转变显著提升治疗彻底性与有效性,为开发新一代协同肿瘤治疗平台提供新思路。
---
2.5 PVP 修饰 RhPd‑H 纳米颗粒的生物界面工程:抗污、长循环与主动靶向行为
RhPd‑H 纳米颗粒优异的体内分布行为与其表面性质介导的一系列生物学效应密切相关。BCA 蛋白吸附实验证实,聚乙烯吡咯烷酮(PVP)修饰使材料在血清与肿瘤间质液中均具备显著抗污能力,有效抑制蛋白冠形成(图 4A)。该特性源于 PVP 链通过空间位阻与氢键构建的致密水化层,可特异性阻断关键调理素及其他介导免疫识别的血浆蛋白吸附。这种分子水平隐身特性直接决定后续细胞相互作用。
为动态评估细胞摄取,将纳米颗粒用罗丹明 B(RB)标记,在 0、2、4、6 h 通过共聚焦显微镜定量内吞情况(图 S8;图 4D)。结果显示巨噬细胞与 4T1 肿瘤细胞摄取动力学差异显著:巨噬细胞在所有时间点吞噬量均极低,荧光强度持续处于低水平,成功实现免疫逃逸;而 4T1 肿瘤细胞则呈快速、时间依赖性的纳米颗粒累积。共聚焦图像定量分析显示该差异随时间逐渐明显,6 h 时荧光强度为巨噬细胞的 2 倍(图 4E),为细胞偏好性提供动态视觉证据。
为直接比较生理竞争条件下肿瘤细胞与各类免疫细胞的摄取偏好,构建两种精准共培养模型并行分析。在“全竞争模型”中,4T1 肿瘤细胞与等量外周血白细胞共培养。流式细胞术分析显示,与 RB 标记的 RhPd‑H 纳米颗粒共孵育后,4T1 细胞平均荧光强度(MFI)约为总免疫细胞群的 6 倍(图 4B)。
为更精准研究循环中主要吞噬细胞的作用,构建 4T1 细胞与中性粒细胞等量共培养的靶向竞争模型。值得注意的是,尽管中性粒细胞与嗜酸性粒细胞是免疫细胞中活性最强的吞噬亚群,其纳米颗粒摄取效率仍显著低于 4T1 肿瘤细胞。上述结果表明,该纳米颗粒不仅可逃逸包括中性粒细胞、单核/巨噬细胞在内的关键吞噬系统清除,还能被肿瘤细胞主动高效内吞(图 4C)。这种同时实现免疫逃逸与肿瘤靶向偏好的独特细胞行为,是实现高效肿瘤靶向递送的核心机制。
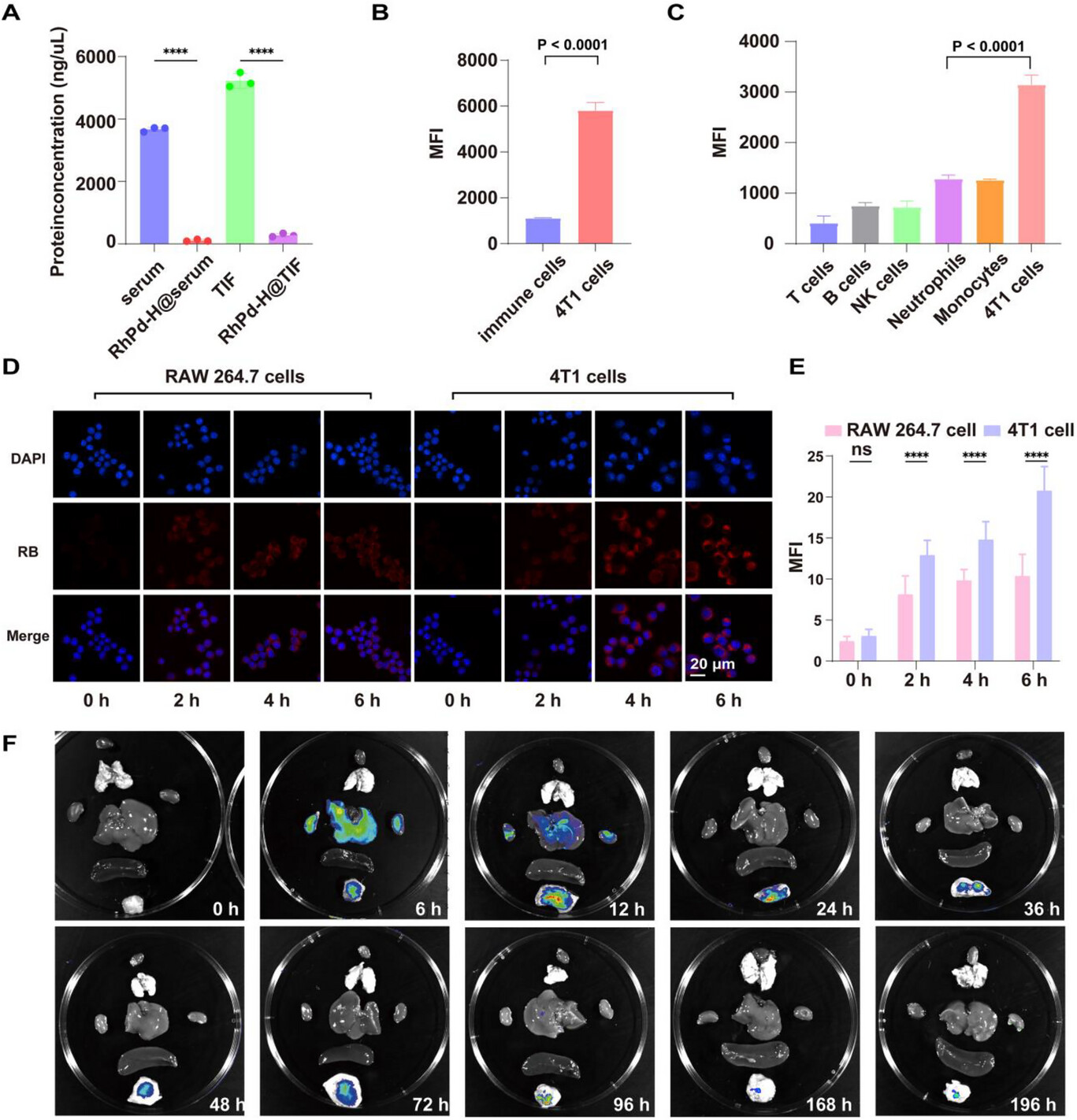
图 4
RhPd‑H的抗污性能、免疫逃逸与肿瘤靶向能力。(A) 血清与肿瘤间质液中RhPd‑H表面蛋白吸附量定量分析(n=3)。(B) 外周血总免疫细胞与4T1细胞对RhPd‑H摄取能力对比(n=4)。(C) 各类免疫细胞与4T1细胞对RhPd‑H摄取能力对比(n=4)。(D, E) 不同时间(0、2、4、6 h)下,罗丹明B(RB)标记的RhPd‑H与RAW 264.7巨噬细胞、4T1细胞共孵育的共聚焦激光扫描显微镜(CLSM)荧光图像(D)及定量结果(E)(n=25)。(F) 离体荧光成像显示RB标记的RhPd‑H在小鼠体内的分布与滞留情况。采用双侧Student’s t检验进行统计学分析:*p < 0.05,**p < 0.01,***p < 0.001,****p < 0.0001。
4T1细胞摄取效率的显著提升,可归因于其本身较高的内吞活性与PVP表面修饰的协同作用。由于巨噬细胞与中性粒细胞主要依赖调理素介导的吞噬作用,PVP包覆可有效逃逸其识别与清除。同时,肿瘤细胞通常高表达B类清道夫受体(SR‑B)等内吞受体,能够在低蛋白冠状态下直接识别纳米颗粒。高浓度PVP表面修饰可有效抑制非特异性蛋白吸附;此外,形成的亲水性高分子层可通过优化空间构象与界面相互作用,主动促进纳米颗粒与受体的结合。共培养实验明确证实,这种细胞偏好性并非单纯源于培养条件差异,而是在两种细胞共存的更接近生理环境中,肿瘤细胞所具备的稳定竞争优势。这种良好的细胞靶向偏好对于提高纳米颗粒的肿瘤靶向生物利用度至关重要,也是其体内优异分布特征的细胞学基础。
上述结果充分证实,肿瘤细胞对纳米颗粒的高效摄取,是其体内肿瘤富集量高、滞留时间长的关键机制(图4F)。RhPd‑H纳米颗粒在肿瘤部位表现出极为持久的滞留效果,注射后信号可检测长达196 h。这种超长肿瘤滞留性源于强大的协同机制:免疫逃逸带来的长效体循环,随后被肿瘤细胞主动高效内吞。这一“渗透至肿瘤间质—被肿瘤细胞主动捕获”的两步过程,为纳米颗粒构建了难以被清除的屏障,使其在肿瘤组织内长期驻留。
与之相对,其全身安全性与高效清除特性同样值得关注。24 h内,心脏、肝脏、脾脏、肺脏、肾脏等主要正常器官的荧光信号均降至可忽略水平,表明材料可在体内快速、彻底地清除。这种“肿瘤长期滞留+全身快速清除”的独特体内分布特征,对治疗效果与生物安全性均具有重要意义。从安全性角度,纳米颗粒在重要器官内无长期蓄积,显著降低脱靶毒性与潜在慢性损伤风险,这也是多数纳米药物普遍存在的问题。兼具长效肿瘤驻留与快速体内清除的特性,充分体现了本研究PVP修饰RhPd‑H纳米颗粒优异的体内性能与临床转化潜力。
---
2.6 体内治疗效果评价
在体内治疗评价中(图5A),将荷4T1肿瘤小鼠随机分为6组:PBS组、RhPd组、RhPd‑H组、PBS+近红外(NIR)组、RhPd+NIR组与RhPd‑H+NIR组。当肿瘤体积约为100 mm³时,以5 mg/kg的统一剂量经尾静脉注射相应制剂。注射24 h后,近红外照射组的肿瘤部位采用808 nm激光(1 W/cm²,5 min)进行照射,并于48 h后重复照射以强化治疗效果。
值得注意的是,由于激光照射会触发RhPd‑H部分释氢,我们评估了该治疗条件下其类过氧化物酶(POD)活性是否受影响。经过两轮激光照射后,RhPd‑H仍保持较强的类POD活性,仅略有下降,且显著高于RhPd组(图S9)。这种持续的类POD活性保障了RhPd‑H在本治疗方案下的疗效。
在14天内每两天监测一次肿瘤体积与小鼠体重,以评估治疗效果与全身毒性。治疗结果显示,联合治疗组的抗肿瘤效果呈现明显的逐级增强趋势(图5B、C、E)。如预期所示,单独使用RhPd或RhPd‑H而不进行激光照射的组别,与PBS对照组相比仅表现出微弱的肿瘤生长抑制,说明在无外源激活条件下,纳米颗粒的催化活性不足以实现有效抑瘤。
为进一步探究光热效应,我们利用红外热像仪监测近红外照射过程中的肿瘤温度,RhPd+NIR组与RhPd‑H+NIR组局部温度分别达到52.9 ℃与53.6 ℃,水平相近(图S10)。RhPd+NIR组结合了RhPd的光热效应与催化功能,实现了约53%的中等但显著的肿瘤抑制率。该结果凸显了局部热疗的关键作用:不仅可直接杀伤肿瘤细胞,还能加速催化反应动力学,从而提升治疗效果。
代表三重联合治疗的RhPd‑H+NIR组展现出最强的抗肿瘤效果,肿瘤抑制率高达91%。值得注意的是,尽管RhPd‑H的长效肿瘤滞留性理论上可支持更多次治疗,但我们刻意将方案限制为两次照射。这种精简且有效的方案在实现最优疗效的同时,最大限度降低了对瘤周正常组织的潜在安全风险。
其优异疗效可从机制上归因于氢气带来的治疗策略根本性转变。与单纯光热‑催化治疗直接但短暂的氧化冲击不同,引入的氢气主动重编程线粒体功能,将其从ATP生产器转变为持续产生内源性超氧阴离子的发生器。由此构建了双重持续性活性氧(ROS)攻击:胞质外源性羟基自由基(·OH)直接爆发,与线粒体来源的持续性内源性超氧阴离子(·O₂⁻)协同作用。这种持久的氧化风暴远超细胞自身修复能力,造成不可逆损伤,实现优异的肿瘤清除效果。
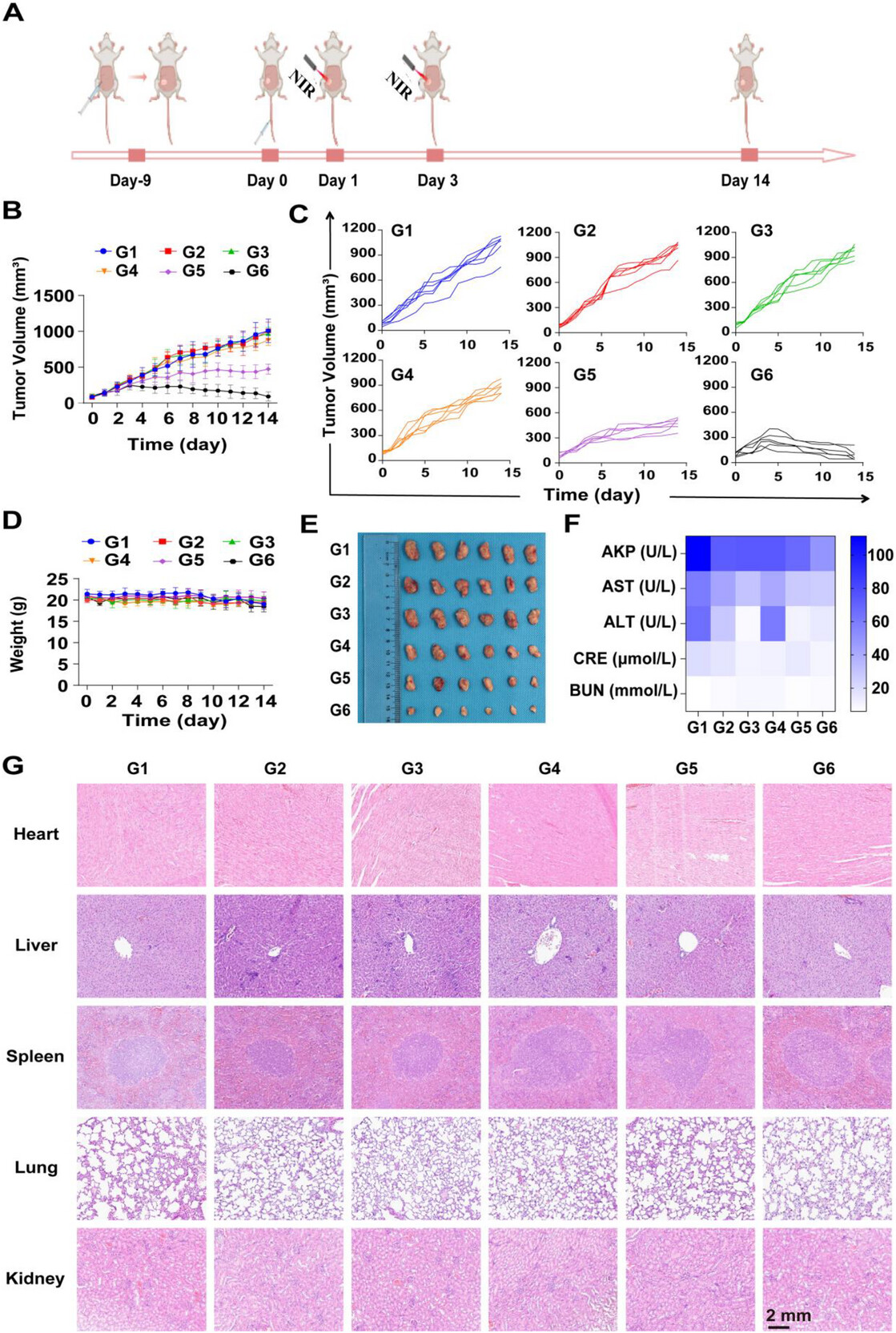
图5 RhPd‑H的体内治疗效果与生物安全性评价
(A) 实验时间线与治疗方案示意图;(B) 治疗期间监测的肿瘤生长曲线(n=6);(C) 不同处理后4T1肿瘤的个体生长曲线;(D) 治疗过程中小鼠体重变化(n=6);(E) 各组剥离肿瘤的代表性照片(n=6);(F) 不同处理组小鼠血液学与生化指标热图;(G) 主要器官的H&E染色切片。
处理组:G1,PBS;G2,RhPd;G3,RhPd‑H;G4,PBS+NIR;G5,RhPd+NIR;G6,RhPd‑H+NIR。
统计学分析采用双侧Student’s t检验:*p < 0.05,**p < 0.01,***p < 0.001,****p < 0.0001。
重要的是,这种强效治疗效果并未引发明显的全身毒性。治疗期间各组小鼠体重均保持稳定,无显著波动(图5D)。主要器官(心、肝、脾、肺、肾)的组织学分析(H&E染色)进一步证实其安全性,未观察到明显病理损伤或病变迹象(图5G)。
该高生物安全性与此前药代动力学结果一致:纳米颗粒在24 h内从所有主要器官快速清除,有效避免了长期器官蓄积,最大程度降低脱靶毒性风险。这种“在肿瘤部位发挥治疗作用后快速清除”的“打了就跑”特征,是纳米药物极为理想的性能。此外,治疗后全面的血液生化分析显示,碱性磷酸酶(AKP)、丙氨酸氨基转移酶(ALT)、天冬氨酸氨基转移酶(AST)、血尿素氮(BUN)与肌酐(CRE)等肝肾功能关键指标均维持在正常生理范围内(图5F),充分保证了本治疗策略的良好生物相容性。
---
2.7 RhPd‑H纳米平台逆转免疫抑制并激发全身抗肿瘤免疫
为阐明光热、催化与氢气联合治疗强效抑瘤的免疫学基础,我们利用流式细胞术系统分析了肿瘤免疫微环境。与PBS对照组及RhPd+NIR组相比,RhPd‑H+NIR处理显著促使巨噬细胞向抗肿瘤M1型极化(图6A、B,圈门策略见图S11)。M1型肿瘤相关巨噬细胞(TAMs,CD80⁺)的平均荧光强度(MFI)较PBS组提升1.99倍,较RhPd+NIR组提升1.53倍。相应地,RhPd‑H+NIR组M1/M2比值升至1.589,显著高于PBS组(0.786)与RhPd+NIR组(1.014),表明氢气强化了抗肿瘤免疫微环境。
氢气释放还显著促进树突状细胞(DCs)成熟。肿瘤引流淋巴结中CD80与MHC‑II的平均荧光强度较PBS组分别提升约1.5倍与1.2倍,较RhPd+NIR组分别提升1.36倍与1.22倍(图6C、D,圈门策略见图S11),揭示氢气在促进抗原呈递与DC介导的T细胞启动中的关键作用。
这种增强的天然免疫激活进一步转化为适应性抗肿瘤免疫。RhPd‑H+NIR处理显著提升肿瘤浸润T细胞比例,其中CD4⁺ T细胞占总T细胞的50.3%,显著高于PBS组(28.9%)与RhPd+NIR组(47.3%);CD8⁺ T细胞占比为43.5%,对应对照组分别为34.3%与43.2%(图6E,圈门策略见图S12)。
CD4⁺ T细胞的活化水平持续增强,CD44与CD69表达较PBS组分别提升约1.3倍与1.45倍,较RhPd+NIR组分别提升1.4倍与1.5倍(图6F)。同样,CD8⁺ T细胞的CD44与CD69表达较PBS组分别上调1.59倍与1.5倍,较RhPd+NIR组分别提升1.33倍与1.67倍(图6G)。
综上,上述结果凸显了氢气在扩增抗肿瘤免疫广度与强度中的不可或缺作用,实现了天然免疫重编程向强效、持久T细胞介导应答的转化。
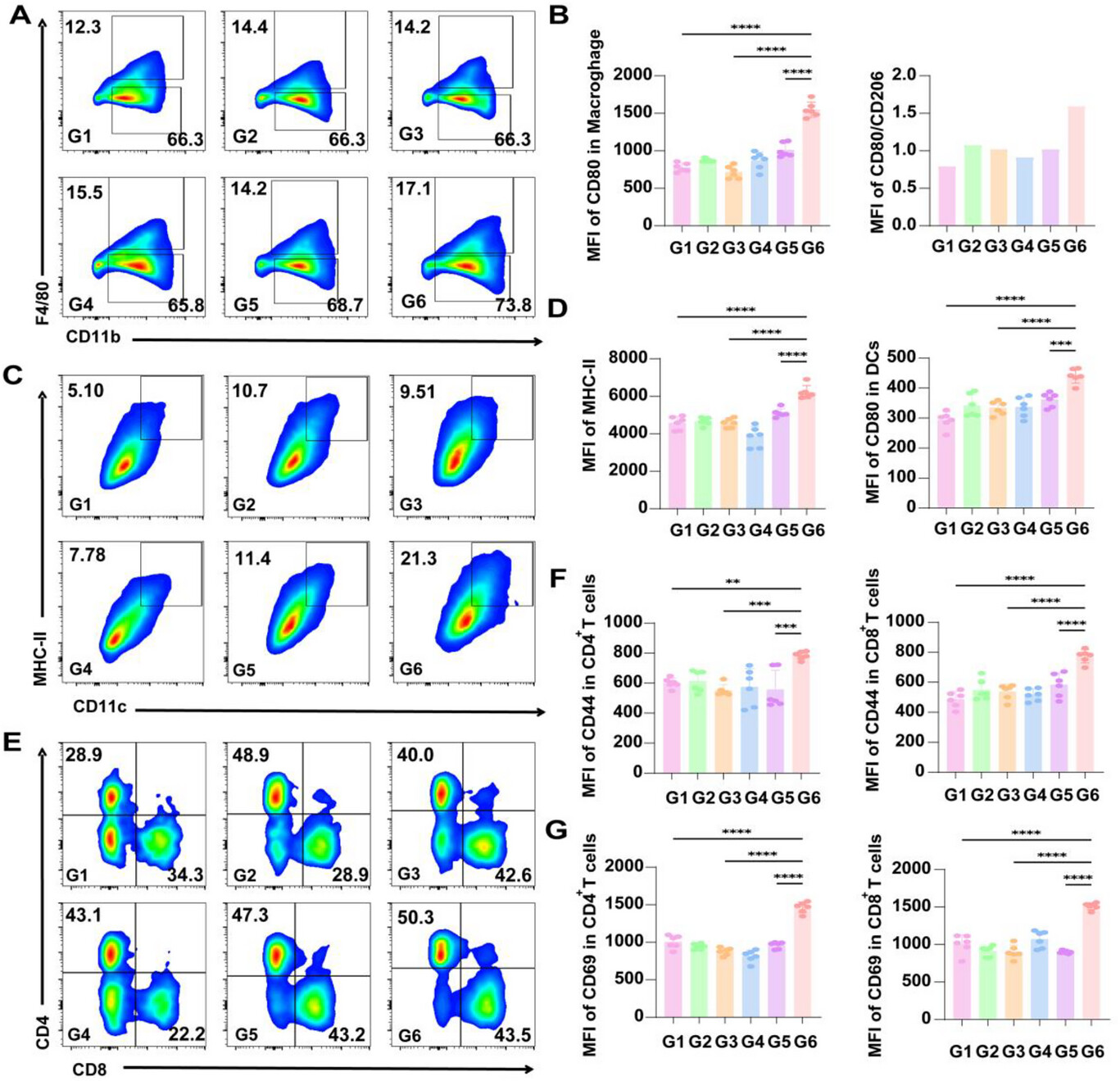
图6 肿瘤特异性免疫应答激活
(A) 不同处理组F4/80⁺/CD11b⁺细胞的代表性流式点图;(B) 巨噬细胞相关标志物CD80表达量及CD80/CD206比值(n=6);(C) 不同处理组MHC‑II⁺/CD11c⁺细胞的代表性流式点图;(D) DCs相关标志物MHC‑II与CD80表达量;(E) 不同处理组CD4⁺/CD8⁺细胞的代表性流式点图;(F) CD4⁺ T细胞中T细胞活化标志物CD44与CD69表达定量分析;(G) CD8⁺ T细胞中CD44与CD69表达量(n=6)。
分组:G1,PBS;G2,RhPd;G3,RhPd‑H;G4,PBS+NIR;G5,RhPd+NIR;G6,RhPd‑H+NIR。
统计学分析采用双侧Student’s t检验:*p < 0.05,**p < 0.01,***p < 0.001,****p < 0.0001。
除肿瘤微环境局部免疫激活外,我们在全身免疫器官脾脏中观察到T细胞活化标志物CD44显著上调(图S13,圈门策略见图S14)。该结果表明,RhPd‑H+NIR处理激发的抗肿瘤免疫应答不仅局限于肿瘤部位,还扩展至全身水平,进一步提示抗肿瘤T细胞可能向效应表型分化,启动了全身性免疫应答。
总而言之,RhPd‑H纳米平台通过促进DC成熟、增强T细胞浸润与活化、重编程巨噬细胞极化,局部逆转免疫抑制微环境;并通过预先激活脾脏T细胞,诱导初步全身免疫活化。局部与全身效应协同,显著提升抗肿瘤免疫应答的强度与持久性,为其在肿瘤免疫治疗中的后续应用奠定坚实基础。
---
3 结论
综上,本研究成功构建了一种新型近红外响应型RhPd‑H纳米平台,整合纳米催化治疗、光热转换与氢气释放功能。该纳米平台具备良好的肿瘤靶向能力与生物利用度,表现为肿瘤长效滞留与正常器官快速清除的特征,保证了高治疗特异性与最小化全身毒性。
更重要的是,与传统催化或光热治疗依赖短暂、一次性活性氧爆发不同,本平台可触发持续、自放大的氧化风暴。这种双重来源的氧化风暴由光热增强催化产生的外源性·OH,与氢气重编程线粒体持续产生的内源性·O₂⁻共同驱动。从急性氧化冲击向持续性氧化攻击的转变,有效突破细胞抗氧化能力上限,导致肿瘤细胞不可逆死亡。
此外,这种强效治疗效果可触发强烈的免疫原性细胞死亡(ICD),激活树突状细胞并启动全身抗肿瘤免疫应答,使RhPd‑H成为一体化联合治疗平台。因此,该平台通过代谢干预与免疫活化协同,显著提升催化治疗效果,为构建以持续性氧化破坏为核心的先进催化治疗提供全新思路。
尽管RhPd‑H平台展现出良好治疗效果,但本研究仍存在一定局限性。目前结果主要基于皮下肿瘤模型,无法完全模拟深部或转移灶的微环境;此外,纳米材料的长期生物安全性与潜在蓄积毒性仍需在未来研究中进行更系统的探究。
---
4 实验方法
4.1 试剂
乙酰丙酮钯(II)(98%)购自Sigma‑Aldrich公司,乙酰丙酮铑(III)(97%)购自中国阿拉丁公司。亚甲基蓝(MB)、罗丹明B(RB)购自中国上海阿拉丁公司。DCFH‑DA、DHE、MitoSOX Red、JC‑1试剂盒、DiO、CCK‑8购自北京索莱宝科技有限公司。胎牛血清(FBS)购自Gibco BRL公司(美国卡尔斯巴德)。所有实验用水均为超纯水。所有试剂均直接使用,未经进一步纯化。
4.2 RhPd‑H光热转换性能评价
RhPd‑H与RhPd的详细合成方法(包括掺氢过程及相关表征)已在我们前期工作中报道。
在三种实验条件下评估RhPd‑H的光热性能:
1. 对不同浓度(20、40、60、80、100 µg/mL)的RhPd‑H溶液采用808 nm激光(1 W/cm²)照射5 min,利用红外热像仪记录温度变化;
2. 对40 µg/mL的RhPd‑H样品采用不同功率密度(0.8、1.0、1.2 W/cm²)的808 nm激光照射5 min,实时监测温度变化;
3. 对40 µg/mL样品进行5次反复加热‑冷却循环以评估光热稳定性,每轮循环为激光照射5 min(808 nm,1 W/cm²)后自然冷却20 min,热成像仪持续追踪温度。
4.3 过氧化物酶活性检测
在pH 6.0条件下,将含TMB(0.4 mM)、H₂O₂(50 mM)与RhPd‑H纳米酶(50 µg/mL)的混合液进行孵育,同时设置不含纳米酶的TMB与H₂O₂对照组。在652 nm处测定各组吸光度,评估RhPd‑H的类POD活性。
为探究pH对催化活性的影响,将反应物浓度固定(TMB 0.4 mM,H₂O₂ 50 mM,RhPd‑H 50 µg/mL),调节体系至不同pH(5.0、6.5、7.4),监测652 nm处吸光度变化,分析纳米酶在不同酸度下的催化性能。
此外,为考察纳米酶浓度对催化活性的影响,在pH 6.0条件下,将不同浓度(25、50、100 µg/mL)的RhPd‑H与TMB(0.4 mM)、H₂O₂(50 mM)共孵育,测定652 nm处吸光度,系统分析催化反应随纳米酶浓度的变化趋势。
4.4 密度泛函理论(DFT)计算
所有DFT计算均采用Vienna Ab Initio Simulation Package(VASP)软件中的平面波基组进行,电子与离子相互作用采用投影缀加波(PAW)势描述。交换关联作用采用广义梯度近似(GGA)下的Perdew‑Burke‑Ernzerhof(PBE)泛函计算。平面波截断能为500 eV,残余力与能量收敛标准分别设为0.05 eV/Å与10⁻⁵ eV。采用Grimme方法的经验校正(DFT‑D3)描述范德华相互作用。布里渊区采用Monkhorst‑Pack方法进行2×2×1 k点网格采样。
采用计算氢电极(CHE)模型获取尿素电化学合成各可能步骤的吉布斯自由能变化(ΔG)。所有电化学步骤的∆G定义为:
ΔG = ΔE + ΔEZPE − TΔS
其中反应能(ΔE)直接由DFT总能分析得到;产物与反应物之间的零点能差(ΔEZPE)由振动频率计算得到;ΔS为室温(T=298.15 K)下产物与反应物的熵变。
4.5 释氢动力学检测
配制0、3.125、6.25、12.5、25、50 µg/mL的MB溶液,测定664 nm处吸光度,建立标准曲线。
为评估光触发释氢行为,将500 µL RhPd‑H(1 mg/mL)与MB(终浓度50 µg/mL)混合于12孔板,采用808 nm激光(1 W/cm²)照射,利用紫外‑可见分光光度计实时监测664 nm处吸光度。
对于释氢动力学,将相同体系在808 nm激光(1 W/cm²)下进行3次开关循环(照射5 min,休息5 min),连续记录吸光度变化,根据标准曲线定量释氢量。
肉眼验证实验:将100 µL RhPd‑H(1 mg/mL)加入96孔板,经808 nm激光(1 W/cm²)照射5 min后,取10 µL置于载玻片,显微镜观察气泡生成情况。
为获得不同功率密度下的温升与释氢曲线,将500 µL含MB(50 µg/mL)的RhPd‑H(1 mg/mL)分别采用0、0.3、0.6、0.9、1.2、1.5 W/cm²的808 nm激光照射10 min,记录溶液温度并持续测定664 nm吸光度至稳定,根据标准曲线计算释氢量。
4.6 4T1细胞与免疫细胞共培养体系的RhPd‑H摄取实验
将4T1细胞以1×10⁵个/孔的密度接种于12孔板,孵育24 h后用PBS洗3次去除血清。加入按1:40000稀释的羧基荧光素琥珀酰亚胺酯(CFSE),37 ℃染色10 min。染色后用PBS洗3次,胰酶消化收集细胞,离心获得细胞沉淀。
从Balb/c小鼠采集外周血,采用50 µL肝素钠抗凝,重悬4T1细胞,制备含4T1细胞与白细胞的混合悬液。
将细胞悬液与RB标记的RhPd‑H(40 µg/mL)按1:1体积比(各100 µL)混合,在金属浴中37 ℃、500 rpm孵育1 h,每20 min轻轻晃动一次。
孵育结束后,450×g离心5 min收集所有细胞,洗去未被吞噬的纳米材料,重悬于含2%胎牛血清的PBS(F‑PBS)。加入流式染色染料,室温染色15 min,加入1 mL 2% F‑PBS终止染色,450×g离心5 min,沉淀重悬于200 µL PBS,过滤后进行流式细胞术分析。
4.7 细胞水平氢气介导生物还原作用评价
4T1肿瘤细胞在含10% FBS与1%青霉素‑链霉素的RPMI‑1640完全培养基中培养,培养条件为37 ℃、5% CO₂。
以1×10⁴个/孔的密度接种于96孔板,培养24 h后进行处理。每孔加入100 µL含300 µg/mL MB的完全培养基,孵育1 h。吸除培养基,用PBS洗3次,每孔加入100 µL含80 µg/mL RhPd或RhPd‑H的完全培养基,对照组仅加入PBS。
经808 nm激光(1 W/cm²)照射5 min后,显微镜观察MB还原导致的蓝色褪去现象。
4.8 RhPd‑H@血清蛋白冠复合物的制备与表征
将RhPd‑H溶液(1 mg/mL)与健康人血清按1:3体积比混合,37 ℃孵育1.5 h。孵育结束后,4 ℃、14000 rpm离心10 min收集复合物沉淀,小心弃去上清,用预冷PBS洗涤3次,保留最后一次洗涤上清用于后续分析。最终将沉淀重悬于预冷PBS,得到RhPd‑H@血清蛋白冠复合物。
蛋白定量:取10 µL复合物与200 µL BCA工作液(A液:B液=1:50)混合,37 ℃孵育30 min,测定562 nm处吸光度(A562)。
4.9 RhPd‑H@肿瘤抗原蛋白冠复合物的制备与表征
采用RPMI‑1640完全培养基将RhPd‑H稀释至50 µg/mL。4T1细胞与RhPd‑H共孵育4 h后,近红外激光照射处理组(1 W/cm²,10 min),随后放回培养箱过夜孵育。收集培养基,4 ℃、14000 rpm离心10 min去除细胞与碎片,保留上清,采用BCA法测定蛋白含量。
将原始RhPd‑H纳米颗粒与所得上清按预设比例混合,37 ℃孵育1.5 h。4 ℃、14000 rpm离心10 min,沉淀用冰PBS洗涤3次,最终重悬于冰PBS,得到RhPd‑H@肿瘤抗原蛋白冠复合物。采用BCA法定量复合物蛋白含量,初步表征蛋白冠组成。
4.10 细胞毒性检测
将4T1与MCF‑7细胞以1×10⁴个/孔的密度接种于96孔板,培养24 h后进行处理。吸除原培养基,更换为含0、5、10、20、40、60 µg/mL RhPd或RhPd‑H的完全培养基。孵育4 h后,近红外处理组采用808 nm激光(1 W/cm²)照射5 min,所有组别继续孵育20 h。
弃去上清,PBS轻柔洗涤一次,每孔加入100 µL含10% CCK‑8试剂的完全培养基,37 ℃避光孵育1.5 h,酶标仪测定450 nm处吸光度。
4.11 细胞摄取实验
制备RB标记的RhPd‑H(RhPd‑H@RB):将0.5 mL RhPd‑H(0.5 mg/mL)与1 mg RB(溶于0.5 mL超纯水)混合,室温200 rpm振荡24 h。通过反复离心洗涤去除未结合染料,至上清无荧光信号。
将4T1与RAW 264.7细胞培养至对数生长期,以1×10⁵个/孔接种至含爬片的12孔板,过夜孵育。每孔加入RhPd‑H@RB至终浓度40 µg/mL,继续孵育4 h。
吸除上清,PBS轻柔洗涤一次,加入适量DiO染色工作液,37 ℃孵育30 min。弃去上清,PBS轻柔洗涤一次,4%多聚甲醛室温固定10 min,PBS再次洗涤。小心取出爬片,用含DAPI的抗荧光淬灭封片液封片,室温避光晾干后共聚焦显微镜成像。
4.12 细胞内活性氧水平检测
将4T1细胞以1×10⁴个/孔接种于96孔板,培养24 h。吸除旧培养基,分为6组:PBS、RhPd、RhPd‑H、PBS+NIR、RhPd+NIR、RhPd‑H+NIR。各组分别用PBS、RhPd(80 µg/mL)或RhPd‑H(80 µg/mL)处理4 h,PBS洗去未结合材料。近红外组采用808 nm激光(1 W/cm²)照射5 min。
每孔加入10 µL含DCFH‑DA的培养基(DCFH‑DA按1:1000稀释于RPMI‑1640空白培养基),轻柔混匀,37 ℃孵育20 min完成染色。PBS洗去未结合染料,在0~240 min内使用酶标仪测定荧光强度(激发488 nm,发射525 nm)。
4.13 细胞爬片法4T1细胞共聚焦成像
简要步骤:12孔板每孔滴加PBS,放入无菌爬片。以1×10⁵个/孔接种4T1肿瘤细胞,恒温培养箱培养24 h。吸除旧培养基,分为6组:PBS、RhPd、RhPd‑H、PBS+NIR、RhPd+NIR、RhPd‑H+NIR。加入相应溶液处理4 h,PBS洗去未结合材料。近红外组采用808 nm激光(1 W/cm²)照射5 min,放回培养箱继续处理。
随后进行三种染色:
1. 2 h后,每孔加入100 µL含DCFH‑DA(1:1000稀释于1640培养基)与MitoSOX Red(1:1000稀释于1640培养基)的培养基,37 ℃染色20 min。
2. 加入0.5 mL JC‑1工作液,37 ℃染色20 min。
3. 加入100 µL含DHE(1:1000稀释于1640培养基)的培养基,37 ℃染色20 min。
染色结束后吸除上清,预冷无菌PBS轻柔漂洗爬片细胞两次,加入预冷4%多聚甲醛固定液完全覆盖爬片,室温固定15 min。去除固定液,PBS洗涤3次。载玻片滴加含DAPI核染的封片液,无菌镊子小心取出爬片,细胞面朝下贴于载玻片中央,避免气泡。晾干后共聚焦显微镜观察成像。
4.14 肿瘤细胞内ATP水平检测
检测经PBS、RhPd+NIR、RhPd‑H+NIR处理的4T1细胞内ATP水平。以1×10⁵个/孔接种于12孔板,培养24 h。更换含PBS、RhPd或RhPd‑H(80 µg/mL)的新鲜培养基,继续孵育4 h。近红外组采用808 nm激光(1 W/cm²,5 min)照射,放回培养箱。
在照射后1 h与3 h提取ATP:每孔加入相当于培养基体积1/10的ATP裂解液,反复吹打裂解细胞。裂解液12000×g离心5 min(4 ℃),收集上清用于检测。
ATP检测:96孔板每孔加入100 µL ATP检测工作液,室温平衡5 min。加入20 µL样品或ATP标准品,立即混匀,延迟2 s后采用发光仪测定相对发光单位(RLU)。
4.15 RhPd‑H在小鼠体内的组织分布
6~8周雌性BALB/c小鼠左侧乳腺脂肪垫皮下接种4T1细胞(2×10⁶个/只)建立肿瘤模型。当肿瘤体积约100 mm³时,经尾静脉注射RB标记的RhPd‑H,剂量为5 mg/kg。
在注射后指定时间点处死小鼠,收集心脏、肝脏、脾脏、肺脏、肾脏等主要器官及肿瘤组织,对RB荧光信号进行离体成像与分析,评估RhPd‑H的时空分布。
4.16 体内治疗效果评价
按照4.15所述方法构建荷瘤小鼠模型。所有动物实验经吉林大学实验动物伦理委员会批准(批准号:KT202406002)。
当肿瘤体积约100 mm³时,小鼠随机分为6组:(1) PBS组;(2) RhPd组;(3) RhPd‑H组;(4) PBS+NIR组;(5) RhPd+NIR组;(6) RhPd‑H+NIR组。经尾静脉注射相应制剂,剂量5 mg/kg。
注射24 h后,近红外组肿瘤部位采用808 nm激光(1 W/cm²)照射5 min,48 h后进行第二次照射。每两天监测一次肿瘤体积与小鼠体重。第14天处死所有小鼠,绘制肿瘤生长曲线评估治疗效果。
4.17 生物安全性评价
采集小鼠血液进行全血细胞分析。随后处死小鼠,摘取心脏、肝脏、脾脏、肺脏、肾脏等主要器官及肿瘤组织。称量完整肿瘤组织重量并拍照。所有组织样本经4%多聚甲醛固定24 h,石蜡包埋、切片、H&E染色,光学显微镜观察组织形态结构变化,评估治疗方案的安全性。
4.18 流式细胞术样品制备
将肿瘤组织剪碎,在消化缓冲液中37 ℃、100 rpm振荡消化35 min。冰上轻柔过70 µm细胞筛,所得细胞悬液重悬于含2% FBS的PBS,450×g离心5 min。沉淀重悬于6~10 mL 35% Percoll分离液,22 ℃、400×g离心25 min,升降速均设为0。弃去上清,细胞沉淀重悬于含2% FBS的PBS,转移至离心管。加入600 µL红细胞裂解液处理5 min,PBS终止裂解,离心。沉淀重悬于250 µL含2% FBS的PBS。
取50 µL细胞悬液与0.5 µL Fc阻断剂4 ℃孵育10 min,随后按表面标志物染色方案加入荧光偶联抗体,室温避光孵育15~20 min。染色后用含2% FBS的PBS洗涤,450×g离心5 min,最终沉淀重悬于200 µL PBS,过滤后流式分析。
取约1/3脾脏组织,机械研磨过70 µm细胞筛获得单细胞悬液。450×g离心5 min,重悬于250 µL含2% FBS的PBS。取2/5细胞悬液与Fc阻断剂4 ℃孵育10 min,抗体染色40 min。加入500 µL红细胞裂解液处理10 min,1 mL含2% FBS的PBS终止反应。离心后沉淀重悬于200 µL PBS,过滤后流式分析。
4.19 数据统计学分析
采用GraphPad Prism软件绘制图表。实验数据以平均值±标准差表示。采用双侧Student’s t检验判断显著性,p<0.05为具有统计学差异。图中星号代表显著性水平:*p < 0.05,**p < 0.01,***p < 0.001,****p < 0.0001。
https://blog.sciencenet.cn/blog-41174-1529875.html
上一篇:氢气吸入对脑血流的影响大【新进展】
下一篇:重新认识一种隐藏的膳食化合物
