博文
最大的癌症全基因组测序研究  精选
精选
||
最大的癌症全基因组测序研究
诸平

据《科学家》(The Scientist)杂志网站2024年4月17日报道,最大的癌症全基因组测序研究(The Largest Whole-genome Sequencing Study in Cancer)。13880例肿瘤的全面基因组测序揭示了可能影响患者治疗和预后的体细胞和种系突变(somatic and germline mutations)。科学家开发了一种生物信息学管道(bioinformatics pipeline),用于整合来自13880位肿瘤患者的全测序数据和患者临床数据1。他们发现了生存和治疗反应的基因组生物标志物(genomic biomarkers)。
癌症没有单一的基因蓝图。相反,每一种癌症都利用了一系列获得性突变(acquired mutations),这些突变赋予细胞选择性优势,以及优越的免疫逃避和增殖策略。由于新一代测序技术,许多被诊断患有特定癌症的患者可以发现他们的肿瘤是否含有特定的突变,使他们更容易受到特定治疗的影响。然而,靶向方法无法捕获患者肿瘤复杂遗传结构中的全套改变和生物标志物,这可能会使个体患者的最佳治疗方案变得模糊。
在《自然医学》(Nature Medicine)上发表的一项研究中,研究人员开发了一种生物信息学管道(bioinformatics pipeline),用于整合来自13880名肿瘤患者的全基因组测序(whole-genome sequencing简称WGS)数据和匹配的患者临床数据1。这项大规模研究揭示了影响预后的体细胞和种系DNA突变,强调了综合癌症基因组学(comprehensive cancer genomics)对患者预后的潜在影响。
马克·考菲尔德(Mark Caulfield,Fig. 1)现在是伦敦玛丽女王大学(Queen Mary University of London)的基因组医学研究者,他曾是英国基因组学公司(Genomics England)的首席科学家,负责监督10万人基因组计划(100,000 Genomes Project | Genomics England)。2019年,他因在该项目上的贡献被授予爵士爵位(knighthood)。
伦敦玛丽女王大学的基因组医学研究员、该研究的合著者马克·考菲尔德说:“癌症是一种基因组紊乱的疾病。”出现的许多基因突变与癌症的生长无关,但其他突变携带的信息可以让研究人员预测肿瘤的进展和脆弱性。马克·考菲尔德和他的团队开始寻找这些信息丰富的基因指纹。
对于许多癌症来说,通过基因组检测来识别特定的基因变异,比如单核苷酸多态性(single-nucleotide polymorphisms),已经是标准治疗的一部分。然而,这些目标面板的范围是有限的。没有参与这项研究的美国密歇根大学(University of Michigan)癌症研究者阿鲁尔·钦奈扬(Arul Chinnaiyan)说:“(综合方法)可以让你全面了解单个患者的肿瘤发生了什么,如果你最终只使用目标面板,你可能会错过这些。这项研究确实体现了这一点的重要性。”
为了探索影响健康和疾病的基因,英国政府于2013年建立了10万基因组计划( 100,000 Genomes Project)2,由政府所有的英国基因组公司运营,研究人员开始在英国公共资助的医疗保健系统——国家医疗服务体系(National Health Service简称NHS)内对患者的基因组进行测序。在该倡议的癌症项目中,研究人员和临床医生合作确定驱动癌症进展的基因变化。在目前的研究中,研究人员对患者的肿瘤及其匹配的生殖系DNA进行了测序。分析患者的正常遗传DNA和肿瘤DNA可以提供对其他癌症易感性的更全面的了解(can provide a bigger picture),或者告诉临床医生,在任何影响药物代谢的基因变异的情况下,哪种治疗方法更适合患者3 。
13880个样本涵盖了33种肿瘤类型。在一个单一的测试中,WGS可以检测体细胞小变异,包括单核苷酸变异(single nucleotide variants简称SNV)和插入及缺失,以及拷贝数畸变(copy number aberrations简称CNA)。通过整合纵向的、真实世界的临床数据,研究人员可以评估这些基因组因素(genomic factors)在治疗反应和生存中的作用。阿鲁尔·钦奈扬说:“整个研究的范围令人印象深刻。”
可以参看精准医学:癌症治疗的新时代(Precision Medicine: A New Era in Cancer Therapy)。
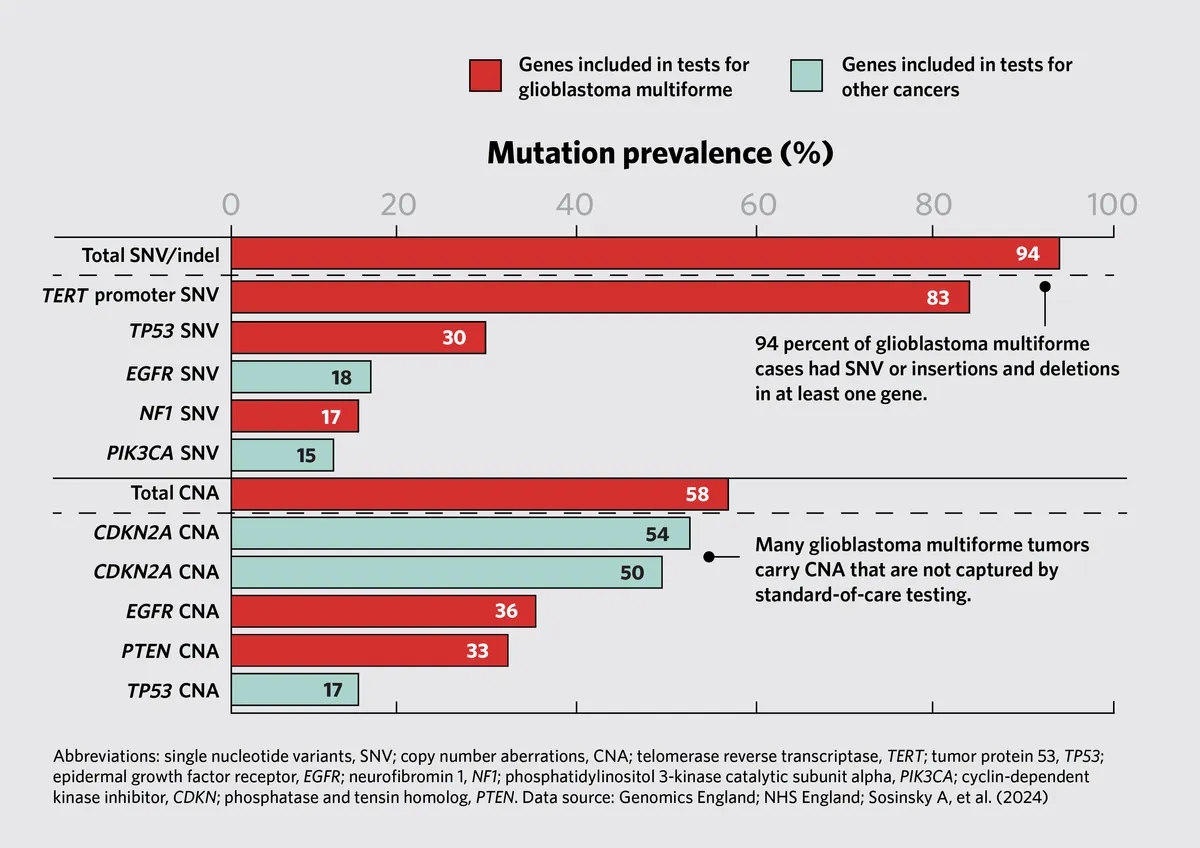
马克•考菲尔德和他的团队首先研究了最新版本的国家癌症基因组测试目录(National Genomic Test Directory for Cancer简称NGTDC)中包含的基因突变率,该目录根据患者的特定癌症类型指定了哪些基因组组符合条件。马克•考菲尔德说:“研究结果非常惊人。”例如,他们发现94%的多形性胶质母细胞瘤(glioblastoma multiforme),一种侵袭性脑癌患者的肿瘤在至少一个基因中有小变异,58%的肿瘤在至少一个基因中含有CNA。标准的、有针对性的检测小组不能检测到许多观察到的突变。马克•考菲尔德说:“这种复杂的基因结构得出的结论是,要真正表征神经胶质瘤的基因变异,一个完整的基因组将是最好的测试。”
在各种癌症类型中,最常发生突变的基因是肿瘤蛋白53 (tumor protein 53简称TP53)。然而,在单个癌症类型中,每种突变的发生都是可变的,突出表现为TP53 SNV在卵巢高级别浆液性癌(ovarian high grade serous carcinoma)中的高频率(90%)和间皮瘤(mesothelioma)中的低频率突变(7%)。
全基因组测序捕获胶质母细胞瘤多形性遗传复杂性(glioblastoma multiforme genetic complexity)。这些数据是基于国家癌症基因组测试目录(National Genomic Test Directory for Cancer)中显示的测试基因。该图2(Fig. 2)包含了SNV和CNA在癌症类型中的前5个突变率。
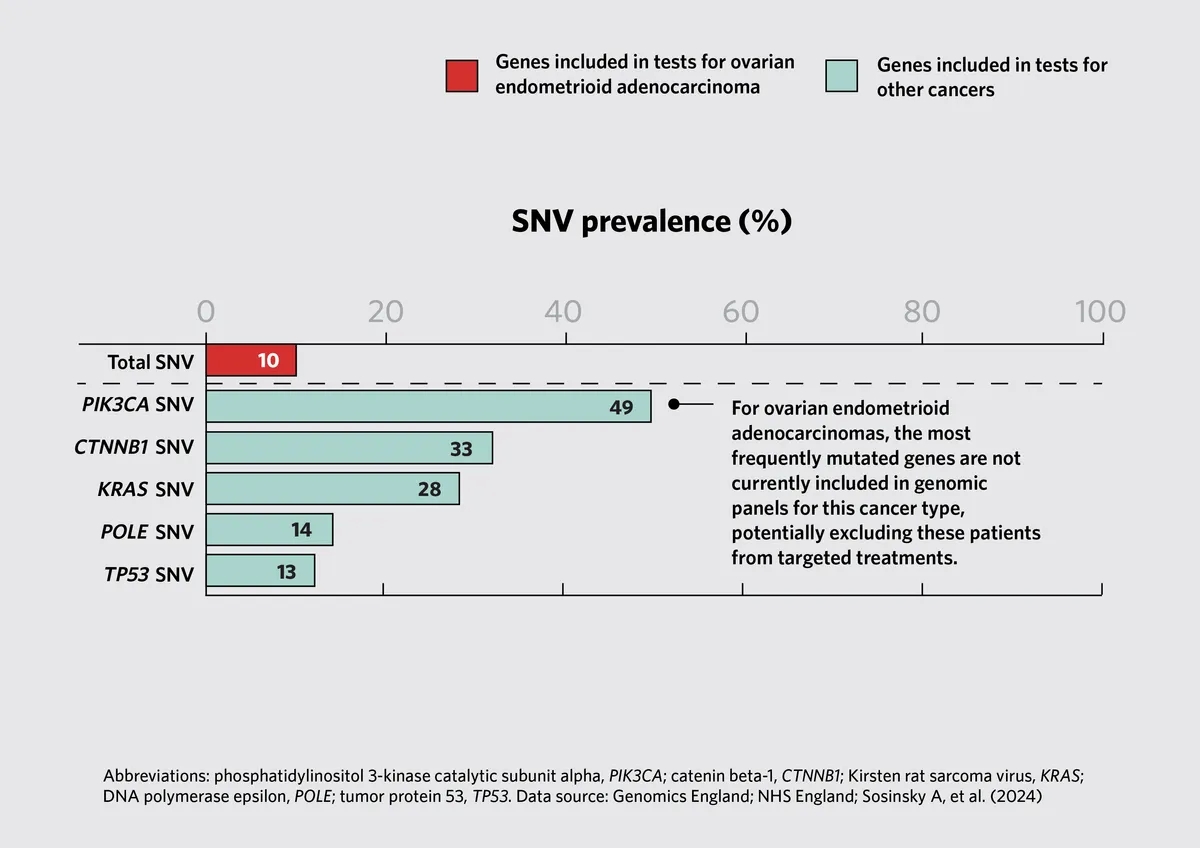
研究小组还评估了未包括在癌症类型标准治疗测试方案中的基因的突变率。例如,54%的子宫体子宫内膜癌(uterine corpus endometrial carcinomas)和49%的卵巢子宫内膜样腺癌(ovarian endometrioid adenocarcinomas)携带一种磷脂酰肌醇3-激酶催化亚单位α (phosphatidylinositol 3-kinase catalytic subunit alpha简称PIK3CA)突变。然而,在英国的医疗保健系统中,这种突变只在乳腺浸润性癌(breast invasive carcinomas)中进行了测试。这些发现表明,科学家们可以在这些癌症的临床试验中测试PIK3CA抑制剂的治疗潜力。
上述图3(Fig. 3):全基因组测序揭示卵巢子宫内膜样腺癌的可操作突变。这些数据是基于国家癌症基因组测试目录中显示的测试基因。该图像包括癌症类型的前五种单核苷酸变异(single nucleotide variants简称SNV)。
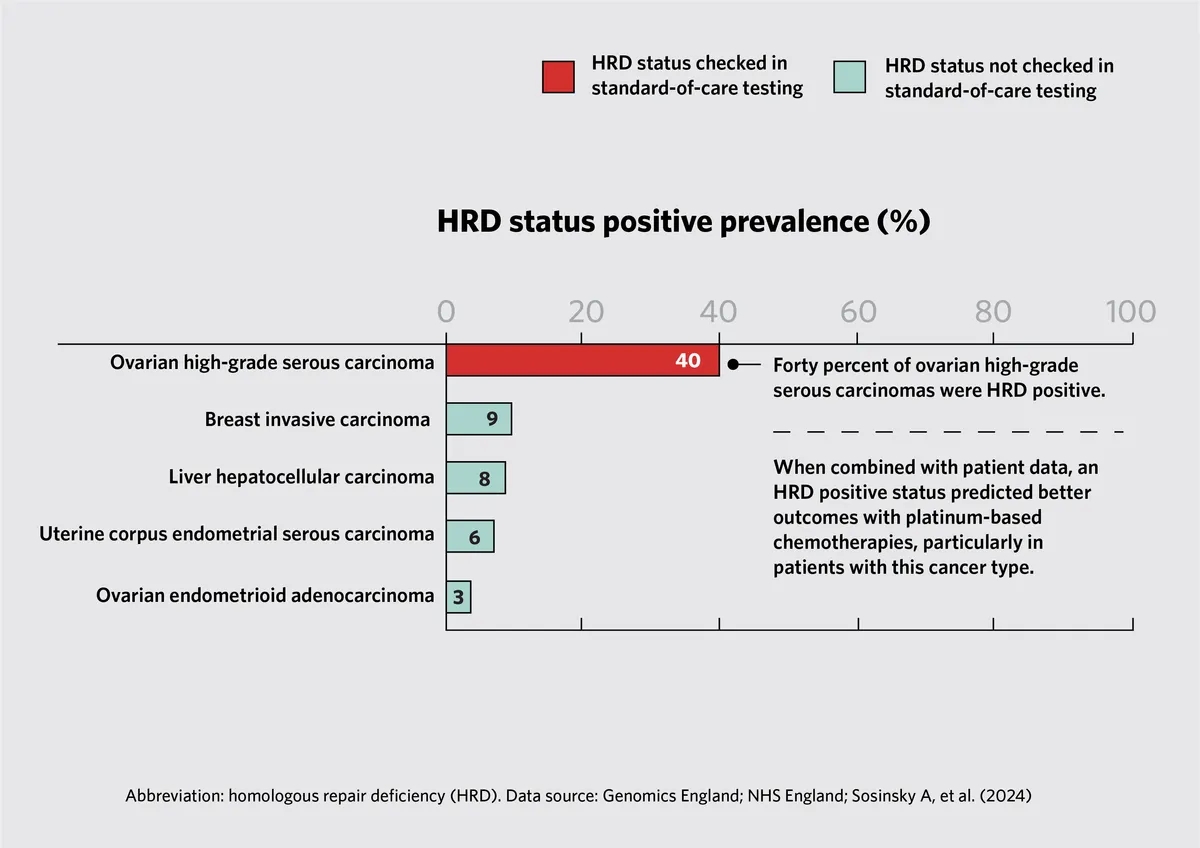
同源重组缺陷(Homologous recombination deficiency简称HRD)是指细胞无法通过同源重组修复DNA中的双链断裂。马克•考菲尔德和他的团队发现40%的卵巢高级别浆液性癌有HRD基因。当他们将患者临床数据纳入分析时,他们发现,相对于HR通路基因没有遗传改变的患者,具有高HRD率的肿瘤患者在铂类化疗(platinum-based chemotherapies)中获得了更好的结果;对于高级别卵巢癌患者尤其如此。
图4(Fig. 4)是泛基因组标记提供预测性生物标记。这些数据是基于国家癌症基因组测试目录(National Genomic Test Directory for Cancer)中显示的测试基因。
由于研究小组使用了一种综合的WGS方法,他们可以对患者肿瘤和非肿瘤DNA进行串联测序,以寻找携带有关患者恶性肿瘤易感性信息的变异,从而指导他们的临床管理。例如,13%的高级别浆液性卵巢癌患者具有BRCA1和BRCA2基因变异。这种癌症类型的患者如果携带易感生殖系变异,就会有更早的癌症发病。马克•考菲尔德说:“如果我们能在有强烈癌症病史的家庭中利用这些信息,也许我们就能更好地进行筛查,并将筛查重点放在特定的个体身上。”
目前的研究只包括WGS数据,因此该管道只涉及患者的基因组图谱。阿鲁尔•钦奈扬说:“包括RNA测序或转录组测序将会很有吸引力,因为这给了你一个肿瘤微环境的表观遗传框架。他们缺少了RNA测序带来的基因组的功能方面。”
无论如何,他指出基因组的发现是令人印象深刻和有趣的。“[这项研究]将对癌症研究界产生影响,”阿鲁尔•钦奈扬说,他的研究小组已经获得了支持正在进行的研究的数据(access to the data)。
当10万基因组计划于2013年启动时,癌症的基因检测是有限的。WGS彻底改变了癌症研究,揭示了癌细胞复杂的遗传结构;然而,这项技术在很大程度上仅限于研究。
马克•考菲尔德说:“未来的癌症治疗几乎肯定会涉及到某种程度的基因组分析,或者可能是多组学分析(multi-omics),这将使我们能够更正确地为患者选择治疗方法。”阿鲁尔•钦奈扬同样认为WGS将在未来的癌症治疗中发挥重要作用。阿鲁尔•钦奈扬说:“精确肿瘤学(precision oncology)的长期愿景是在成本可控的情况下实施这种综合方法。”尽管自人类基因组计划以来,对整个人类基因组进行测序的成本已经大幅下降,但一旦考虑到处理、分析、解释和存储成本,其成本仍然高(prohibitively expensive)得令人望而却步。4,5
然而,马克•考菲尔德希望这一天能快点到来。他说,“最终,整个基因组的价格将会下降,到那时,很可能每个人都将转向全基因组测序,因为它将具有成本效益。这将使我们能够发现人们癌症中超出我们目前知识范围的不寻常特征。”
马克•考菲尔德说,下一阶段是将WGS与临床试验结合起来,以了解它们是否可以识别治疗反应的生物标志物。有了肿瘤的突变结构和电子临床数据,研究人员可以在临床试验结束后跟踪患者的健康历程。
上述介绍,仅供参考。欲了解更多信息,敬请注意浏览原文或者相关报道。
1. Sosinsky A, et al. Insights for precision oncology from the integration of genomic and clinical data of 13,880 tumors from the 100,000 Genomes Cancer Programme. Nat Med. 2024;30(1):279-289.
2. Turnbull C, et al. The 100,000 Genomes Project: Bringing whole genome sequencing to the NHS. BMJ. 2018; 361: k1687.
3. Kurian A W, et al. Germline genetic testing after cancer diagnosis. JAMA. 2023; 330(1): 43-51.
Akhoundova D, Rubin MA. The grand challenge of moving cancer whole-genome sequencing into the clinic. Nat Med. 2024; 30(1): 39-40.
4. Schwarze K, et al. The complete costs of genome sequencing: A microcosting study in cancer and rare diseases from a single center in the United Kingdom. Genet Med. 2020; 22(1): 85-94.
https://blog.sciencenet.cn/blog-212210-1431030.html
上一篇:科学家解决了几十年的显微镜问题
下一篇:老科学,新转折:古青蛙化石颠覆了百年信仰
