博文
第一性原理计算初学者的福音-Materials project资源
|
第一性原理计算是凝聚态物理、化学、材料研究领域的一个重要手段。对于初学者来说,这个领域的入门还是有一定的难度的。其中,一个较大的困难是计算参数的设置。另外一个困难是计算结果的合理性分析与理论分析。
当然,对于计算参数的设置,我这里介绍一个非常有效的参考资料。即所谓的材料界的谷歌:加州伯克利大学劳伦斯实验室及麻省理工学院等单位组建的Materials project网站(https://www.materialsproject.org/)。该网站的强大,我们后续会介绍。在此,我重要介绍其对第一性原理初学者的参数设置的帮助。
该网站估计把几乎所有的晶体材料都做了一个系统计算。我们进入该网站之后,
第一步、用自己的email地址注册一下;
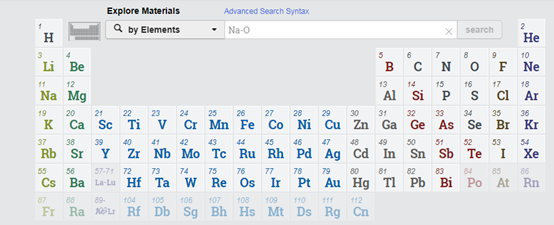
第二步、根据元素组合搜索相应的化合物。在下面的元素周期表内用鼠标点击想要的元素就可以了。注意设置搜索条件:by Elements。如下图所示。
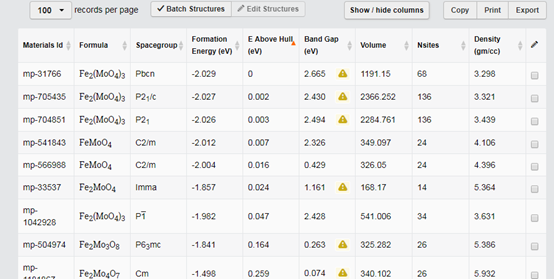
第三步、点击搜索之后会在元素周期表下方出现一些结构。如下图所示:
第四步、选择想要的化合物,鼠标点击,可以显示三维结构。
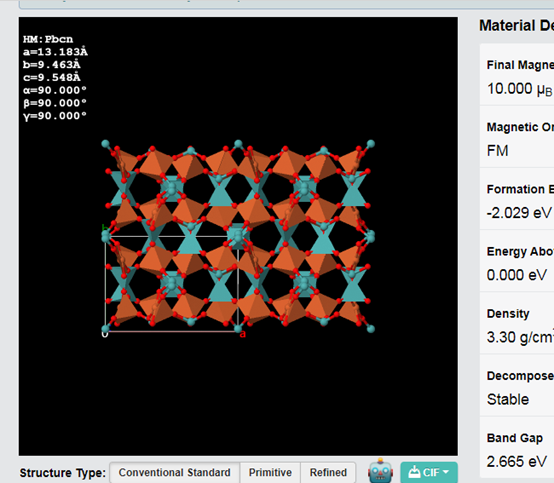
注意,图片下面有CIF,点击可以下载结构的cif文件。可以在Materials studio中直接打开。
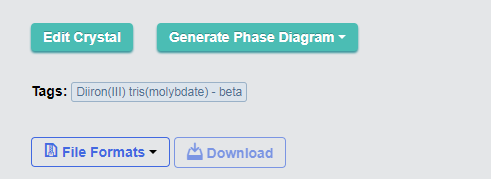
第五步、结构下方有File Formats和Download标志。点击File Formats,选择VASP,再点击右边的Download就可以下载VASP计算的相应文件了。里面有INCAR、KPOINTS、POSCAR、POTCAR。初学者可以好好看看,参考一下他们的设置方法。
该网站包含了几乎所有的晶体材料的模拟计算。所以,对于第一性原理计算的初学者来说,你可以到里面找到相应的结构与计算参数设置方法,降低入门门槛。当然,由于该网站把几乎所有的晶体材料都做了一个基本性质的计算。因此,以后想要通过算一些晶体结构的基本物理化学性质而发表学术论文则是一件比较难的事了。这也需要材料模拟人员深入使用这个数据库,更深层次的开发新材料、设计研究内容。
https://blog.sciencenet.cn/blog-65253-1162938.html
上一篇:“怕老婆”是男人的屈辱吗?--论“问所欲为”心态
下一篇:二维磁性材料的压电系数计算经验分享