博文
多吃容易导致糖尿病,根源竟然在大脑!  精选
精选
|
多吃容易导致糖尿病,根源竟然在大脑!
最新研究发现,多吃带来的代谢紊乱,根源在大脑不是外周。这似乎和最新减肥药的作用靶点在大脑,产生的效应在外周类似。
肥胖人群患糖尿病的风险是瘦人的十倍。试图理解这一现象的研究人员在驱动身体战斗或逃跑反应的同一系统中找到了答案。这些发现挑战了长期以来关于过量饮食如何使你生病的假设。
研究表明,高脂饮食会触发全身神经递质的激增,导致肝脏中脂肪组织的快速分解——这一过程通常由胰岛素的释放来控制。高水平游离脂肪酸的释放与一系列健康状况有关,从糖尿病到肝功能衰竭。
研究人员此前认为,肥胖引起的糖尿病的主要问题是胰岛素活性失常,这意味着身体无法阻止危险脂肪酸的释放。但最新的研究发现,“不是刹车失灵”,而是有另一个杠杆——肝脏和其他组织中的神经递质——正在大力推动油门,奥地利格拉茨大学的生物化学家Martina Schweiger说。“这确实是一个范式转变。”
该研究于10月21日发表在《细胞代谢》杂志上。
胰岛素抵抗
全球超过8.9亿人患有肥胖症,这是发展为糖尿病和其他代谢紊乱的主要风险因素。研究人员早就知道,当胰岛素停止降低血糖水平时,疾病就会进展。新泽西州新布朗斯维克罗格斯大学的生理学家Christoph Buettner和Kenichi Sakamoto及其同事希望更好地理解这种胰岛素抵抗的本质。
Buettner长期研究胰岛素在大脑中调节代谢的作用,因此他和他的团队将注意力转向交感神经系统,该系统向全身组织传递诸如去甲肾上腺素等神经递质。研究人员使用了他们之前开发的一种小鼠模型,其中删除了一个表达产生这些神经递质所需关键酶的基因。这个基因仅在小鼠的四肢和一些器官中被删除,以确保它能存活。
研究人员给经过修改的小鼠提供了富含猪油、椰子油和大豆油的高脂饮食。在超过两个月的观察期间,无论是经过修改的还是未经修改的小鼠都摄入了同样多的食物,体重增加相似,并保持了类似的胰岛素信号活动,即胰岛素与其在细胞上的目标受体结合后发生的一系列事件。
但是,经过修改的小鼠并没有出现脂肪组织分解增加和胰岛素抵抗,最终也没有显示出脂肪肝和组织炎症增加的迹象。另一方面,未经修改的小鼠出现了胰岛素抵抗,这可能导致糖尿病。它们还表现出炎症和肝病迹象增加。
大脑中的信号
这些发现表明,神经递质负责驱动胰岛素抵抗及相关问题,Buettner说。他和他的同事们现在正在探索这些神经递质在其他条件下的作用,例如由更年期引起的胰岛素抵抗。
Schweiger说:“这项研究相当扎实”,但“仍然有一些缺失的部分”。例如,现在的问题是高脂饮食如何触发神经递质激增,她说。
她还表示,需要更多的工作来更好地理解这些发现对人们的影响。到目前为止,阻断涉及交感神经系统的神经递质活动的药物在肥胖人群中尚未显示出益处。Buettner说,可能针对这些药物在特定组织中进行靶向治疗,并避免大脑,可能会更有前景。
过度营养通过增加交感神经系统活动导致胰岛素抵抗和代谢紊乱
要点:
高脂饮食(HFD)通过增加交感神经系统(SNS)活动迅速损害胰岛素作用
减少SNS活动可以防止高脂饮食引起的胰岛素抵抗和代谢紊乱
早期高脂饮食在细胞胰岛素信号受损之前就诱导了胰岛素抵抗
脂肪分解是升高的SNS活动引起胰岛素抵抗的关键机制
总结:
肥胖引起的胰岛素抵抗的机制尚未完全了解,因为通常被认为是胰岛素抵抗主要驱动因素的细胞胰岛素信号受损并不总是伴随胰岛素作用的受损。过度营养迅速增加血浆去甲肾上腺素(NE),表明交感神经系统(SNS)过度激活。然而,SNS在肥胖中的作用是有争议的,因为有报道说SNS活动(SNA)既增加又减少。在这里,我们展示了减少SNS释放儿茶酚胺(CA)可以保护免受过度营养引起的胰岛素抵抗以及胰高血糖素血症、脂肪组织功能障碍和脂肪肝疾病,正如我们利用一个可诱导且仅限于外围的酪氨酸羟化酶基因敲除小鼠模型所展示的那样。增强的SNA引起胰岛素抵抗的一个关键机制是通过触发脂肪组织的脂肪分解。增加的SNA成为独立于细胞胰岛素信号之外,过度营养引起的胰岛素抵抗和代谢疾病发病的关键驱动因素。
图表摘要
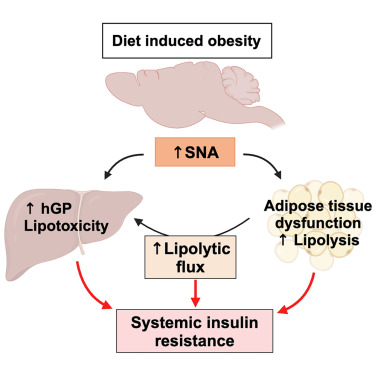
肥胖主要通过诱导胰岛素抵抗引起2型糖尿病和代谢性疾病。与瘦人相比,超重者患糖尿病的风险增加3倍以上,而肥胖者则超过10倍。胰岛素抵抗可以在肥胖显现之前迅速在过度营养期间发展,导致葡萄糖耐受不良、脂肪组织功能障碍和肝脂肪变性。脂肪组织的胰岛素抵抗是脂肪组织功能障碍的关键因素,后者表现为肥大的脂肪细胞,不受控制的脂肪分解,减少的脂肪生成能力,增加的脂肪组织炎症和纤维化。肝脏中的胰岛素抵抗表现为不受控制的肝糖产生(hGP),这是糖尿病空腹高血糖的主要驱动因素。肝脏胰岛素抵抗也被认为是导致肝脂肪变性的原因,这种变性可以进展为与代谢功能障碍相关的脂肪肝疾病(MAFLD)。MAFLD存在于超过70%的所有2型糖尿病病例中,并且是纤维化和肝功能衰竭的最常见原因。
肥胖引起的胰岛素抵抗机制尚未完全了解。根据主流观点,肥胖通过破坏目标组织(如肝脏和脂肪组织)中的细胞内胰岛素信号来引起胰岛素抵抗,这通过经典的胰岛素受体-IRS-磷脂酰肌醇3激酶(PI3K)-AKT-FoxO1信号级联进行。然而,在肥胖的早期阶段,这些组织中的细胞胰岛素信号似乎完好无损,尽管胰岛素无法抑制脂肪分解和hGP。因此,目标组织中受损的细胞胰岛素信号可能无法完全解释整体胰岛素抵抗,定义为胰岛素对葡萄糖和脂肪酸的抑制作用减弱。
另一种可能导致肥胖引起的胰岛素抵抗的机制是增强的反向调节信号,这是由于交感神经系统(SNS)活动(SNA)增加及其主要激素去甲肾上腺素(NE)导致的,它诱导了肾上腺能信号。SNS在肥胖中的作用是有争议的,因为既有增加也有减少SNA的报道。这些矛盾的发现可能归因于用于评估SNA的方法的局限性。通过神经记录、NE周转或血浆NE水平直接测量SNA的研究经常报告肥胖中SNA增加。相比之下,研究肾上腺能信号或效应途径如脂肪组织脂肪分解的研究通常报告由儿茶酚胺(CA)激活减少,这被解释为肥胖中SNA减少。这一矛盾可以通过CA抗性来解释,即由于β-肾上腺素受体表达减少或肾上腺素脱敏,未能对CA刺激产生正常的生理反应,导致第二信使环磷酸腺苷(cAMP)水平降低。CA抗性在肥胖中很常见,并可以由慢性交感神经过度激活引起。
虽然SNS的主要传递物质是NE,但SNS的一个重要部分是释放肾上腺素的肾上腺腺体;SNS还控制着胰腺α细胞释放胰高血糖素。这些激素通过增加hGP和诱导脂肪组织脂肪分解来拮抗胰岛素的作用。据报道,小鼠在高脂饮食(HFD)喂养4周后NE水平会增加,这是一种常用于研究过度喂养导致肥胖和胰岛素抵抗的模型。肾上腺素对肥胖引起的代谢紊乱的贡献是有争议的。尽管HFD喂养据报道会增加小鼠血浆肾上腺素水平,但在肥胖人类中肾上腺素分泌并未增加。普遍认为,高胰高血糖素血症在肥胖引起的胰岛素抵抗中发挥作用,其中胰高血糖素分泌异常增加是由于α细胞功能失调,定义为葡萄糖未能抑制从胰岛α细胞分泌的胰高血糖素。通常认为α细胞功能失调主要是由于β细胞未能在葡萄糖水平升高时增加胰岛素分泌,因为胰岛素会抑制α细胞分泌胰高血糖素。增加的SNA可能在多大程度上导致肥胖引起的高胰高血糖素血症尚不清楚。
为了阐明SNS在过度营养和肥胖引起的胰岛素抵抗及代谢疾病中的作用,我们的研究首先集中在评估过度营养对SNA的影响,然后使用最近开发的外周限制性和可诱导的酪氨酸羟化酶(th)敲除小鼠模型研究SNS的作用。在这些小鼠中,外周组织的交感神经CA水平显著降低,而中枢神经系统(CNS)的CA水平保持不变。与之前的去神经模型不同,这种方法避免了手术或化学去神经可能引起的潜在发育效应或局部炎症。我们的发现揭示了过度营养触发了SNA激增,这是胰岛素抵抗、脂肪组织功能障碍和肝脂肪变性的原因。增加的SNA导致代谢疾病的一个主要机制是增加脂肪组织脂肪分解,这是SNS的一个关键靶点。
https://blog.sciencenet.cn/blog-41174-1457923.html
上一篇:深度睡眠有助于心脏愈合
下一篇:化学家合成了一种“不可能”分子!《科学》
