博文
build tuition之常见概念辨析
||
关注:
1) 电负性与得失电子的关系;
2) 如何有效地进行化学键分析
1. Dipole
Physics A pair of electric charges or magnetic poles, of equal magnitude but of opposite sign or polarity, separated by a small distance.
【物理学】 偶极子:一对电荷或磁极,符号或极性相反但数量相等,彼此隔一定距离
In physics, a dipole is a quantity involved some form of polarity
2. LDOS
Local density of states (LDOS) is a physical quantity that describes the density of states, but space-resolved.
In materials science, this term is useful when interpreting the data from an STM, since this method is capable of imaging electron densities of states with atomic resolution. According to crystal's structure, this quantity can be predicted by computational methods, as for example with density functional theory.
3. Melting and Melting criteria
Melting, or fusion, is a physical process that results in the phase transition of a substance from a solid to a liquid. The internal energy of a substance is increased, typically by the application of heat or pressure, resulting in a rise of its temperature to the melting point, at which the ordering of ionic or molecular entities in the solid breaks down to a less ordered state and the solid liquefies【(使)溶解, (使)液化】.
An object that has melted completely is molten【熔化】. Substances in the molten state generally have reduced viscosity【粘质, 粘性】 with elevated temperature; an exception to this maxim is the element sulfur, whose viscosity increases to a point due to polymerization and then decreases with higher temperatures in its molten state.[1]
Among the theoretical criteria for melting, the Lindemann [4] and Born [5] criteria are those most frequently used as a basis to analyse the melting conditions .
The Lindemann criterion states that melting occurs because of vibrational instability, e.g. crystals melt when the average amplitude of thermal vibrations of atoms is relatively high compared with interatomic distances, e.g. <δu2>1/2 > δLRs, where δu is the atomic displacement, the Lindemann parameter δL ≈ 0.20...0.25 and Rs is a half of the inter-atomic distance.【热运动位移太大,以致ion忘记了怎么回到自己原来的位置--Neil】
The Lindemann melting criterion is supported by experimental data both for crystalline materials and for glass-liquid transitions in amorphous materials. The Born criterion is based on rigidity【坚硬, 僵化, 刻板, 严格, 刚性, 硬度】 catastrophe【大灾难, 大祸】 caused by the vanishing elastic shear modulus, e.g. when the crystal no longer has sufficient rigidity to mechanically withstand load.
Lindemann index
The Lindemann index[1] is a simple measure of thermally driven disorder in atoms or molecules. The local Lindemann index is defined as:[2]
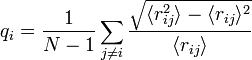
Where angle brackets indicate a time average. The global Lindemann index is a system average of this quantity.
In condensed matter physics a departure from linearity in the behaviour of the global Lindemann index or an increase above a threshold value related to the spacing between atoms (or micelles, particles, globules, etc.) is often taken as the indication that a solid-liquid phase transition has taken place. See Lindemann melting criterion.4.Debye–Waller factor
The Debye–Waller factor (DWF), named after Peter Debye and Ivar Waller, is used in condensed matter physics to describe the attenuation【衰减】 of x-ray scattering or coherent neutron scattering caused by thermal motion.[1][2]
It has also been called the B factor or the temperature factor.【这就是XRD测量中B factor的由来】
Often, "Debye-Waller factor" is used as a generic【 普通的】 term that comprises the Lamb-Mössbauer factor of incoherent neutron scattering and Mössbauer spectroscopy.
The DWF depends on the scattering vector q. For a given q, DWF(q) gives the fraction of elastic scattering;
1 - DWF(q) correspondingly gives the fraction of inelastic scattering. (Strictly speaking, this probability interpretation is not true in general.[3])
In diffraction studies, only the elastic scattering is useful; in crystals, it gives rise to distinct Bragg peaks.
Inelastic scattering events are undesirable as they cause a diffuse background — unless the energies of scattered particles are analysed, in which case they carry valuable information (inelastic neutron scattering).
The basic expression for the DWF is given by
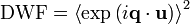
where u is the displacement of a scattering center, and <...> denotes either thermal or time averaging.
Assuming harmonicity of the scattering centers in the material under study, the Boltzmann distribution implies that  is normally distributed with zero mean. Then, using for example the expression of the corresponding characteristic function, the DWF takes the form
is normally distributed with zero mean. Then, using for example the expression of the corresponding characteristic function, the DWF takes the form
![.text{DWF} = .exp.left( -.langle [.mathbf{q}.cdot .mathbf{u}]^2 .rangle .right)](http://upload.wikimedia.org/math/a/9/e/a9ee6e2b1e2c2d53907417603d0760ee.png)
Note that although the above reasoning is classical, the same holds also quantum mechanically.
Assuming also isotropy of the harmonic potential, one may write

where q, u are the magnitudes (or absolute values) of the vectors q, u respectively, and 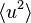 is the mean squared displacement. Note that if the incident wave has wavelength
is the mean squared displacement. Note that if the incident wave has wavelength  , and it is elastically scattered by an angle of
, and it is elastically scattered by an angle of 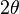 , then
, then
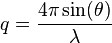
In the context of protein structures, the term B-factor is used. The B-factor is defined as:

It is measured in units of Å2. The B-factors can be taken as indicating the relative vibrational motion of different parts of the structure.
Atoms with low B-factors belong to a part of the structure that is well-ordered. Atoms with large B-factors generally belong to part of the structure that is very flexible.
Each ATOM record (PDB file format) of a crystal structure deposited with the Protein Data Bank contains a B-factor for that atom.
5. Hydrocarbon【烃, 碳氢化合物】
In organic chemistry, a hydrocarbon is an organic compound consisting entirely of hydrogen and carbon.[1] Hydrocarbons from which one hydrogen atom has been removed are functional groups, calledhydrocarbyls.[2] Aromatic hydrocarbons (arenes), alkanes, alkenes, cycloalkanes and alkyne-based compounds are different types of hydrocarbons.
The majority of hydrocarbons found on earth naturally occur in crude oil, where decomposed organic matter provides an abundance of carbon and hydrogen which, when bonded, can catenate to form seemingly limitless chains
6. Jahn-Teller效应
The Jahn–Teller effect, sometimes also known as Jahn–Teller distortion, describes the geometrical distortion of molecules and ions that is associated with certain electron configurations.
This electronic effect is named after Hermann Arthur Jahn and Edward Teller, who proved, using group theory, that orbital nonlinear spatially degenerate molecules cannot be stable.[1]
The Jahn–Teller theorem essentially states that any nonlinear molecule with a spatially degenerate electronic ground state will undergo a geometrical distortion that removes that degeneracy, because the distortion lowers the overall energy of the species.
For description of another type of geometrical distortion that occurs in crystals with substitutional impurities see article off-center ions.
Transition metal chemistry
The Jahn–Teller effect is most often encountered in octahedral complexes of the transition metals.[3]
The phenomenon is very common in six-coordinate copper(II) complexes.[4]
The d9 electronic configuration of this ion gives three electrons in the two degenerate eg orbitals, leading to a doubly degenerate electronic ground state. Such complexes distort along one of the molecular fourfold axes (always labelled the z axis), which has the effect of removing the orbital and electronic degeneracies and lowering the overall energy. The distortion normally takes the form of elongating the bonds to the ligands lying along the z axis, but occasionally occurs as a shortening of these bonds instead (the Jahn–Teller theorem does not predict the direction of the distortion, only the presence of an unstable geometry). When such an elongation occurs, the effect is to lower the electrostatic repulsion between the electron-pair on the Lewis basic ligand and any electrons in orbitals with a zcomponent, thus lowering the energy of the complex. If the undistorted complex would be expected to have an inversion centre, this is preserved after the distortion.
In octahedral complexes, the Jahn–Teller effect is most pronounced when an odd number of electrons occupy the eg orbitals. This situation arises in complexes with the configurations d9, low-spin d7 or high-spin d4 complexes, all of which have doubly degenerate ground states. In such compounds the eg orbitals involved in the degeneracy point directly at the ligands, so distortion can result in a large energetic stabilisation. Strictly speaking, the effect also occurs when there is a degeneracy due to the electrons in the t2g orbitals (i.e. configurations such as d1 or d2, both of which are triply degenerate). In such cases, however, the effect is much less noticeable, because there is a much smaller lowering of repulsion on taking ligands further away from the t2g orbitals, which do not point directly at the ligands (see the table below). The same is true in tetrahedral complexes (e.g. manganate: distortion is very subtle because there is less stabilisation to be gained because the ligands are not pointing directly at the orbitals.
The expected effects for octahedral coordination are given in the following table:
| Number of d electrons | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| High spin | w | w | s | w | w | s | ||||
| Low spin | w | w | w | w | s | s |
w: weak Jahn–Teller effect (t2g orbitals unevenly occupied), s: strong Jahn–Teller effect expected (eg orbitals unevenly occupied), blank: no Jahn–Teller effect expected.
The Jahn–Teller effect is manifested in the UV-VIS absorbance spectra of some compounds, where it often causes splitting of bands. It is readily apparent in the structures of many copper(II) complexes.[2] Additional, detailed information about the anisotropy of such complexes and the nature of the ligand binding can be however obtained from the fine structure of the low-temperature electron spin resonance spectra.
7. Peierls transition
A Peierls transition or Peierls distortion is a distortion of the periodic lattice of a one-dimensional crystal.
Atomic positions oscillate so that the perfect order of the 1-D crystal is broken.
Peierls' Theorem states that a one-dimensional equally spaced chain with one electron per ion is unstable.
It was asserted in the 1930s by Rudolf Peierls. It can be proven using a simple model of the potential for an electron in a 1-D crystal with lattice spacing a.
The periodicity of the crystal creates energy band gaps in the E–k diagram at multiples of the value k = π/a (similar to the result of the Kronig–Penney model, which helps to explain the origin of band gaps in semiconductors).
If the ions each contribute one electron, then the band will be half-filled, up to values of k = ±π/2a in the ground state.
Imagine a lattice distortion where every other ion moves closer to one neighbor and further away from the other, the unfavourable energy of the long bond between ions is outweighed by the energy gain of the short bond.
The period has just doubled from a to 2a. In essence, the proof relies on the fact that doubling the period would introduce new band gaps located at multiples of k = π/2a.
This would cause small energy savings, based on the distortion of the bands in the vicinity of the new gaps.
Approaching k = π/2a from the left, the distortion due to the introduction of the new band gap will cause the electrons to be at a lower energy than they would be in the perfect crystal.
Therefore, this lattice distortion becomes energetically favorable when the energy savings due to the new band gaps outweighs the elastic energy cost of rearranging the ions.
Of course, this effect will be noticeable only when the electrons are arranged close to their ground state – in other words, thermal excitation should be minimized.
Therefore, the Peierls transition should be seen at low temperature. This is the basic argument for the occurrence of the Peierls transition, sometimes called dimerization.
Historical background[edit]
Peierls’ discovery gained experimental backing during the effort to find new superconducting materials. In 1964, Dr. William Little of the Stanford University Department of Physics theorized that a certain class of polymer chains may experience a high TC superconducting transition.[2] The basis for his assertion was that the lattice distortions which lead to pairing of electrons in the BCS theory of superconductivity could be replaced instead by rearranging the electron density in a series of side chains. This means that now electrons would be responsible for creating the Cooper pairs instead of ions. Because the transition temperature is inversely proportional to the square root of the mass of the charged particle responsible for the distortions, the TC should be improved by a corresponding factor:

The subscript i represents "ion," while e represents "electron." The predicted benefit in superconducting transition temperature was therefore a factor of about 300.
In the 1970s, various organic materials such as TTF-TCNQ were synthesized.[3] What was found is that these materials underwent an insulating transition rather than a superconducting one. Eventually it was realized that these were the first experimental observations of the Peierls transition. With the introduction of new band gaps after the lattice becomes distorted, electrons must overcome this new energy barrier in order to become free to conduct. The simple model of the Peierls distortion as a rearrangement of ions in a 1-D chain could describe why these materials became insulators rather than superconductors.
Related physical consequences[edit]
Peierls predicted that the rearrangement of the ion cores in a Peierls transition would produce periodic fluctuations in the electron density. These are commonly called charge density waves, and they are an example of collective charge transport. Several materials systems have verified the existence of these waves. Good candidates are weakly coupled molecular chains, where electrons can move freely along the direction of the chains but motion is restricted perpendicular to the chains. NbSe3 and K0.3MoO3 are two examples in which charge density waves have been observed at relatively high temperatures of 145K and 180K, respectively.[4]
Furthermore, the 1-D nature of the material causes a breakdown of the Fermi liquid theory for electron behavior. Therefore, a 1-D conductor should behave as a Luttinger liquid instead. A Luttinger liquid is a paramagnetic one-dimensional metal without Landau quasi-particleexcitations.
8 分子中的原子理论(Atoms in molecules,简称AIM)
分子中的原子理论(Atoms in molecules,简称AIM)是量子化学的一个模型。它基于电子密度标量场的拓扑性质来描述分子中的成键。
除了成键性质之外,AIM 还根据拓扑性质对全空间进行划分,每个区域内正好包含一个原子核,这种区域给出了量子化学上定义原子的一种方式。通过对每一区域内进行积分,可以得到单个原子的一系列性质。
AIM 方法于上世纪60年代由理查德·贝德提出。在过去的几十年里,AIM 逐渐发展成一种用于解决化学体系中的许多问题的理论,其应用的广泛性远非之前提出的各种模型或理论所能及。[1][2]在 AIM 中,原子表现电子密度梯度场中的吸引子,因而可以通过梯度场的局域曲率来进行定义。这种分析方法一般在文献中称为对电子密度的拓扑分析,尽管这个词与数学中的拓扑一词的含义并不相同。
AIM 理论的主要结果包括:
分子可以人为地划分为各个原子的区域。这些区域之间的分界面为电子密度梯度场的零通量面。原子的物理性质,包括原子有效电荷、偶极矩和能量等,可以通过采用适当的算符在原子区域内进行积分而得到;
当且仅当两个原子之间被一个零通量面分隔开,且该零通量面上有一个(3, −1) 临界点时,认为两个原子间存在键。其中临界点指的是电子密度梯度为零的点。(3, −1) 临界点指的是海森矩阵的本征值中有两个负值和一个正值的临界点。其中 3 表示海森矩阵的非零本征值的个数,而−1表示本征值的符号函数之和。这个临界点称为键临界点(bond critical point, BCP)。
换句话说,键临界点就是电子密度标量场上的一阶鞍点。键径由电子密度梯度场中与键临界点相关联的两个核临界点(即原子核所在位置)指向键临界点两条轨线构成。键径上的每一点在垂直于键径的方向上均为电子密度的极大点。
根据电子密度场在键临界点的拉普拉斯的符号,可以把键分为两类:闭壳层相互作用的拉普拉斯为正,而电子共享相互作用的拉普拉斯为负。
分子成键张力可以用键径与连接两原子核的直线之间的夹角来表征,键径偏离直线越远,表明键中的张力越大。
Atoms in molecules
The Quantum Theory of Atoms in Molecules (QTAIM) is a model of molecular and condensed matter electronic systems (such as crystals) in which the principal objects of molecular structure - atoms and bonds - are natural expressions of a system's observable electron density distribution function. An electron density distribution of a molecule is a probability distribution that describes the average manner in which the electronic charge is distributed throughout real space in the attractive field exerted by the nuclei. According to QTAIM, molecular structure is revealed by the stationary points of the electron density together with the gradient paths of the electron density that originate and terminate at these points. QTAIM was primarily developed by Professor Richard Bader and his research group at McMaster University over the course of decades, beginning with analyses of theoretically calculated electron densities of simple molecules in the early 1960s and culminating with analyses of both theoretically and experimentally measured electron densities of crystals in the 90s. The development of QTAIM was driven by the assumption that, since the concepts of atoms and bonds have been and continue to be so ubiquitously useful in interpreting, classifying, predicting and communicating chemistry, they should have a well-defined physical basis.
QTAIM recovers the central operational concepts of the molecular structure hypothesis, that of a functional grouping of atoms with an additive and characteristic set of properties, together with a definition of the bonds that link the atoms and impart the structure. QTAIM defineschemical bonding and structure of a chemical system based on the topology of the electron density. In addition to bonding, AIM allows the calculation of certain physical properties on a per-atom basis, by dividing space up into atomic volumes containing exactly one nucleus, which acts as a local attractor of the electron density. In QTAIM an atom is defined as a proper open system, i.e. a system that can share energy and electron density, which is localized in the 3D space. The mathematical study of these features is usually referred in the literature as charge density topology.
QTAIM rests on the fact that the dominant topological property of the vast majority of electron density distributions is the presence of strong maxima that occur exclusively at the nuclei, certain pairs of which are linked together by ridges of electron density. In terms of an electron density distribution's gradient vector field, this corresponds to a complete, non-overlapping partitioning of a molecule into three-dimensional basins (atoms) that are linked together by shared two-dimensional separatrices (interatomic surfaces). Within each interatomic surface, the electron density is a maximum at the corresponding internuclear saddle point, which also lies at the minimum of the ridge between corresponding pair of nuclei, the ridge being defined by the pair of gradient trajectories (bond path) originating at the saddle point and terminating at the nuclei. Because QTAIM atoms are always bounded by surfaces having zero flux in the gradient vector field of the electron density, they have some unique quantum mechanical properties compared to other subsystem definitions, including unique electronic kinetic energy, the satisfaction of an electronic virial theorem analogous to the molecular electronic virial theorem and some interesting variational properties.
QTAIM has gradually become a method for addressing possible questions regarding chemical systems, in a variety of situations hardly handled before by any other model or theory in Chemistry[1][2][3][4]
Applications[edit]QTAIM is applied to the description of certain organic crystals with unusually short distances between neighboring molecules as observed by X-ray diffraction. For example in the crystal structure of molecular chlorine the experimental Cl...Cl distance between two molecules is 327 picometres which is less than the sum of the van der Waals radii of 350 picometres. In one QTAIM result 12 bond paths start from each chlorine atom to other chlorine atoms including the other chlorine atom in the molecule. The theory also aims to explain the metallic properties ofmetallic hydrogen in much the same way.
The theory is also applied to so-called hydrogen-hydrogen bonds [5] as they occur in molecules such as phenanthrene and chrysene. In these compounds the distance between two ortho hydrogen atoms again is shorter than their van der Waals radii and according to in silicoexperiments based on this theory, a bond path is identified between them. Both hydrogen atoms have identical electron density and are closed shell and therefore they are very different from the so-called dihydrogen bonds which are postulated for compounds such as (CH3)2NHBH3 and also different from so-called agostic interactions.
In mainstream chemistry close proximity of two nonbonding atoms leads to destabilizing steric repulsion but in QTAIM the observed hydrogen hydrogen interactions are in fact stabilizing. It is well known that both kinked phenanthrene and chrysene are around 6 kcal/mol (25 kJ/mol) more stable than their linear isomers anthracene and tetracene. One traditional explanation is given by Clar's rule. QTAIM shows that a calculated stabilization for phenanthrene by 8 kcal/mol (33 kJ/mol) is the result of destabilization of the compound by 8 kcal/mol (33 kJ/mol) originating from electron transfer from carbon to hydrogen, offset by 12.1 kcal (51 kJ/mol) of stabilization due to a H..H bond path. The electron density at the critical point between the two hydrogen atoms is low, 0.012 e for phenanthrene. Another property of the bond path is its curvature.

Another molecule studied in QTAIM is biphenyl. Its two phenyl rings are oriented at a 38° angle with respect to each other with the planar molecular geometry (encountered in a rotation around the central C-C bond) destabilized by 2.1 kcal/mol (8.8 kJ/mol) and the perpendicular one destabilized by 2.5 kcal/mol (10.5 kJ/mol). The classic explanations for this rotation barrier are steric repulsion between the ortho-hydrogen atoms (planar) and breaking of delocalization of pi density over both rings (perpendicular).
In QTAIM the energy increase on decreasing the dihedral angle from 38° to 0° is a summation of several factors. Destabilizing factors are the increase in bond length between the connecting carbon atoms (because they have to accommodate the approaching hydrogen atoms) and transfer of electronic charge from carbon to hydrogen. Stabilizing factors are increased delocalization of pi-electrons from one ring to the other and the one that tips the balance is a hydrogen - hydrogen bond between the ortho hydrogens.
The hydrogen bond is not without its critics. According to one the relative stability of phenanthrene compared to its isomers can be adequately explained by comparing resonance stabilizations.[6] Another critic [7] argues that the stability of phenanthrene can be attributed to more effective pi-pi overlap in the central double bond; the existence of bond paths are not questioned but the stabilizing energy derived from it is
8.aromaticity
In organic chemistry, aromaticity is a chemical property describing the way in which a conjugated ring of unsaturated bonds, lone pairs, or empty orbitals exhibits a stabilization stronger than would be expected by the stabilization of conjugation alone. The earliest use of the term was in an article by August Wilhelm Hofmann in 1855.[1] There is no general relationship between aromaticity as a chemical property and the olfactory properties of such compounds.
Aromaticity can also be considered a manifestation of cyclic delocalization and of resonance.[2][3][4] This is usually considered to be because electrons are free to cycle around circular arrangements of atoms that are alternately single- and double-bonded to one another. These bonds may be seen as a hybrid of a single bond and a double bond, each bond in the ring identical to every other. This commonly seen model of aromatic rings, namely the idea that benzene was formed from a six-membered carbon ring with alternating single and double bonds (cyclohexatriene), was developed by Kekulé (see History section below). The model for benzene consists of two resonance forms, which corresponds to the double and single bonds superimposing to give rise to six one-and-a-half bonds. Benzene is a more stable molecule than would be expected without accounting for charge delocalization.
Theory[edit]

As is standard for resonance diagrams, a double-headed arrow is used to indicate that the two structures are not distinct entities, but merely hypothetical possibilities. Neither is an accurate representation of the actual compound, which is best represented by a hybrid (average) of these structures, which can be seen at right. A C=C bond is shorter than a C−C bond, but benzene is perfectly hexagonal—all six carbon-carbon bonds have the same length, intermediate between that of a single and that of a double bond.
A better representation is that of the circular π bond (Armstrong's inner cycle), in which the electron density is evenly distributed through a π-bond above and below the ring. This model more correctly represents the location of electron density within the aromatic ring.
The single bonds are formed with electrons in line between the carbon nuclei — these are called σ-bonds. Double bonds consist of a σ-bond and a π-bond. The π-bonds are formed from overlap of atomic p-orbitals above and below the plane of the ring. The following diagram shows the positions of these p-orbitals:

Since they are out of the plane of the atoms, these orbitals can interact with each other freely, and become delocalized. This means that, instead of being tied to one atom of carbon, each electron is shared by all six in the ring. Thus, there are not enough electrons to form double bonds on all the carbon atoms, but the "extra" electrons strengthen all of the bonds on the ring equally. The resulting molecular orbital has π symmetry.

芳香性是一种化学性质,有芳香性的分子中,由不饱和键、孤对电子和空轨道组成的共轭系统具有特别的、仅考虑共轭时无法解释的稳定作用。可以将芳香性看作是环状离域和环共振的体现。一般认为在这些体系中的电子,可以自由在由原子组成的环形结构上运动(离域),这些环形结构含有单键和双键相间,离域的结果是环键的键级趋于均化,给予体系稳定作用。
芳香性的理论最初由凯库勒发展出来,是以六元的苯分子为原型建立起来。理论中的苯有两个共振形态,并有单键和双键相间,但理论上的两种苯(环己三烯)衍生物的这类异构体在实际上无法检测或分离出来,苯事实上是这两个异构体的“杂化体”,并且具有不考虑电子离域时无法解释的稳定性。
被分类为芳香性的化合物通常有以下的条件:
给出离域电子形成π键的原子需处于同一个平面;
原子需组成一个环;
组成π键的电子总数需为 4n+2,即不是4的倍数的双数(休克尔规则);
苯就是一个较好的例子,它适合以上所有条件,并且有6个离域电子(即n=1)。有4n+2个π电子的化合物通常都是芳香性的。环丁二烯只有4个离域电子,所以不属于芳香性化合物。这些只有4n个π电子而又具近似平面结构的环状化合物称为反芳香性化合物。
非芳香族的有机物就叫做脂肪族,脂肪族没有芳香族具有的特殊的共振稳定作用。在受到与平面垂直的外磁场作用时,离域的π电子环电流产生的感应磁场可将整个空间划分为屏蔽区与去屏蔽区,可在核磁共振结果上显示出来。
在商业中最重要的芳香化合物就是苯和甲苯,每年产量极高。从石油中得到的苯和甲苯可用来做其他极有用的日用品材料,包括苯乙烯、苯酚、苯胺及尼龙。
https://blog.sciencenet.cn/blog-567091-737609.html
上一篇:科研精神之:“赠人玫瑰,手留余香"-我眼中的导师
下一篇:学而时习之-Molecular orbital theory