博文
翻译:植物泛基因组综述(Plant pan-genomes are the new reference)
|

泛基因组肯定是未来之趋势,虽然不是每个人都能整整泛基因组,但整整某类基因或基因家族是否可以呢?比如,单参考基因组时代,不同物种中,都发了N多基因家族研究的文章?那么,泛基因组时代,能否从这个方面审视基因家族的研究呢?
Main
泛基因组的概念最早是 2005 年在细菌中提出的,对几个菌株的测序显示,核心基因组占 80% ,其余 20% 在至少一个菌株中缺失。然而,在最初的细菌泛基因组工作之后,植物泛基因组的构建花了将近 10 年的时间。部分原因是数据生成的成本,以及高等生物体中基因的存在和缺失变异(PAV)很少。将泛基因组这个术语应用于植物的第一篇研究发表于 2007 年,其描述了水稻和玉米基因组中的短变异区间。然而,由于同一物种的多个个体缺乏精确的全基因组组装,以致当时还不清楚基因的存在和缺失程度。随着 DNA 测序成本降低,获得并比较物种内不同个体的基因组变得可行,并且发展了三种泛基因组装的一般方法(图 1)。第一种方法是整个基因组的组装和比对,即首先从头组装多个基因组,然后进行基因组水平的比对;基于此,又发展出了迭代组装和 PAV 鉴定 的方法,即将来自多个个体的基因组序列与参考基因组比对,没有比对上的序列被添加到泛基因组上;随后,重新将序列与新构建的泛基因组进行比对以获取 PAV 变异。近年来,基于图形的泛基因组组装得到了迅速发展,最终构建的泛基因组涵盖基因组可变和保守部分。
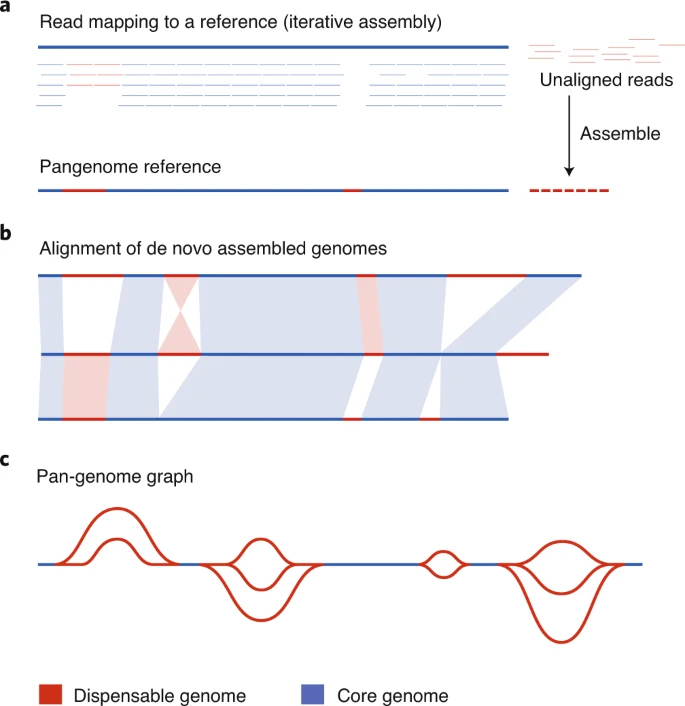
图 1 泛基因组组装方法
前两种方法是高度互补的,整个基因组的组装和比较提供了重要的结构和基因位置信息,而迭代组装方法允许扩展到非常大的个体数量,可以在群体中鉴定稀有基因和PAV. 图形泛基因组已经在细菌中广泛使用,随着近期长读长测序技术的改进,该技术现在才得以在复杂的基因组中变得可行。
每种方法都有自己的优缺点。例如,迭代装配不能区分一个位点的极端序列差异和序列的插入或缺失,而整个基因组装配方法不能区分真正的个体基因组多样性和在装配及注释方法中观察到的常见错误和变异。基于图形的泛基因组有可能代表植物泛基因组学的未来; 然而,对大量高质量长读长测序数据的要求以及大型计算内存的要求,限制了其应用范围,目前其应用仅限于相对较少的个体和较小的基因组。
植物泛基因组研究进展迅速(图2和表1)。首个公布的植物泛基因组是由7个野生大豆基因组构建的。该研究发现了与种子组成、开花和成熟时间、器官大小和生物量有关的可变基因,以及驯化大豆中不存在但在野生大豆中存在的抗病基因。另一项早期的研究对18份拟南芥材料进行了比较,但大部分是比较基因表达和蛋白质亚型,而不是比较基因存在和缺失。早期水稻中的一项研究使用三个分化较大的水稻材料构建了一个小型水稻泛基因组,发现一个品种中 S5杂种不育位点和耐淹水基因 Sub1A 的 PAV 变异。
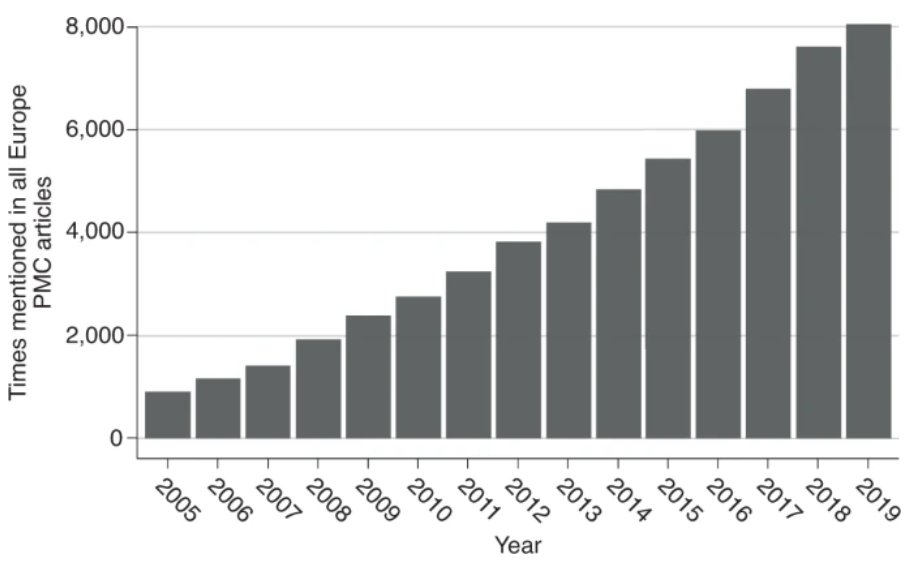
图2 2005-2019年已发表的泛基因组相关文章数量
表1 植物泛基因组研究总结
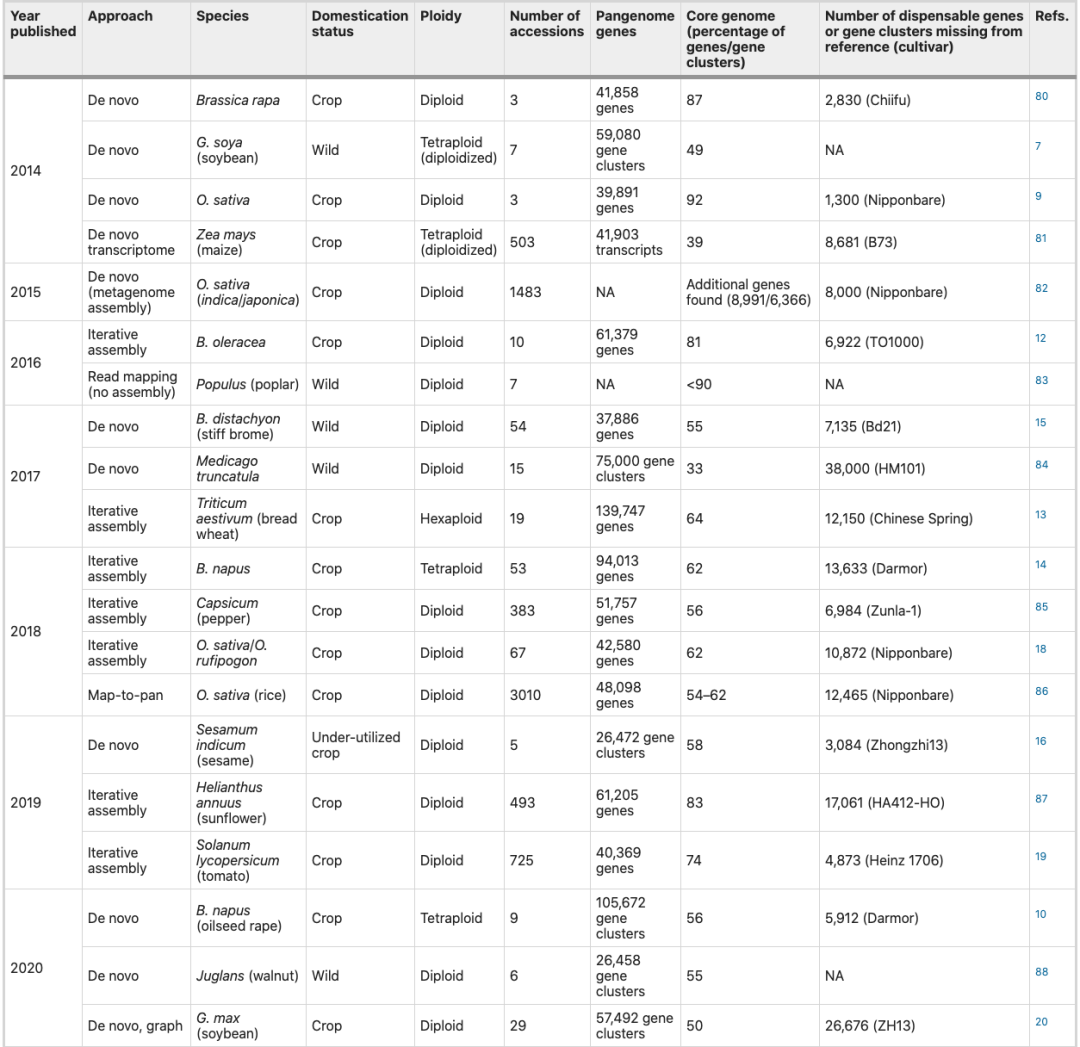
接下来的一项研究比较了8个从头组装的甘蓝型油菜基因组,发现其中两个 hAT 反转录转座子相关的 PAV插入到了已知开花基因中,继而影响了开花时间。在另一项基于七个拟南芥基因组的研究中,发现了与串联重复相关的非共线重排热点(HOTs),进一步分析发现,这些HOTs的减数分裂重组变少,含有的基因也较少,并伴有抗病基因的富集。由于其他物种中缺乏数量众多的高质量基因组,目前不可能在其他植物物种中挖掘HOTs,并且,其与抗病之间的关系仍有待验证。
考虑到生成高质量基因组组装的成本,一些泛基因组研究应用了迭代作图组装方法,包括甘蓝(基于10个个体) ,面包小麦(基于18个个体)和油菜(基于53个个体)。这些早期的植物泛基因组研究产生了两个主要结果: 1)在所研究的每个物种中都有大量的可变基因(15-40%);2) PAV 相关基因往往与生物和非生物胁迫抗性相关。
基于54个短柄草基因组构建的泛基因组,鉴定了7135个可变基因,并发现一些基因虽然在整个群体是可变的,但却是亚群的核心基因,从而促使了种群结构的稳定。利用5个芝麻品种组装了芝麻的泛基因组,对古老和现代芝麻品种进行了基因组比较,结果表明,泛基因组可以帮助追踪在驯化和育种过程中频率发生变化的基因。最近在对基于89个木豆材料为基础构建的泛基因组研究中,利用可变基因进行关联分析,发现3个基因与粒重相关,暗示 PAV 可以作为SNP关联分析的补充。
随着人们对泛基因组研究兴趣的增加,人们对可变基因在群体水平的分布也越来越感兴趣。例如,从1,083份栽培稻和446个野生稻品种中选择66个具有代表性的材料进行分析,发现了10,783个在参考基因组中缺失的基因,这些基因包含以前被报道的重要农艺性状相关基因。一项番茄全基因组研究检测了725个不同品系的变异,确定了4,873个可变基因,其中许多基因与抗病相关,此外,还发现了一个罕见的与番茄风味连锁的等位基因,这个等位基因在驯化过程中被选择,但由于野生渗入,在现代番茄品种中重新出现。最后,在大豆中进行的一项泛基因组研究,即对26个品系进行基因组组装并对2,898个品系进行重测序,进一步发现了与农艺性状相关的位点。
Impact of pan-genomes on plant biology
选择哪个个体的基因组作为参考基因组常常是出于研究历史等方面的考虑。例如,在普通小麦中,来自中国的地方品种中国春的基因组被作为参考基因组使用,这是因为标准细胞核型系统是使用中国春发展起来的。然而,中国春小麦的基因含量与现代品种有很大的不同,第一个面包小麦泛基因组研究发现,18个重新测序的现代品种中有12,150个中国春没有的基因。使用单一的参考基因组可能影响我们对表型遗传基础的理解,例如,小麦抗锈病基因 Lr49在不同品种之间表现出显著的结构变异。使用泛基因组作为参考将拓宽基因组学分析,例如,使用泛基因组提高了二代短reads的比对准确性,从而可获得更高质量的变异和更准确的基因表达量。基于基因数量描述植物物种仍然是一个挑战,特别是考虑到一个物种个体之间的基因 PAV。然而,随着更多物种泛基因组的发展,会促进我们对基因保守和丢失的了解,从而有助于确定物种水平上的基因数量差异。
对基因 PAV 的理解,除基础生物学研究之外,还可以应用于作物改良。作物野生近缘种通常拥有更广泛的基因库,是作物育种中遗传多样性的宝贵来源。二十世纪后期,作物产量增加的30%,可归功于野生近缘种基因在植物育种中的应用。通过泛基因组分析,我们可以研究在驯化和育种过程中基因的保留和丢失情况,继而增加我们对遗传多样性丧失的理解,使基因重新引入现代品种变得可能。例如,西红柿驯化过程中与风味相关的基因丢失,现在又被重新引入现代栽培品种中。对不同环境中野生物种之间的基因PAV进行研究,有助于培育能更好地适应不同环境和气候变化的新品种。
对植物泛基因组的可变基因的功能分析表明,这些基因富集在响应生物和非生物胁迫应答的生物学过程中,特别是存在生物适合度代价的抗病基因。在小麦等单子叶植物以及甘蓝型油菜和番茄等双子叶植物中都观察到了抗病基因PAV,在人类的泛基因组中也有类似发现。这些发现引出了泛 NLR组的概念,即专注于NLR抗病基因的泛基因组学研究。然而,这类研究迄今只在拟南芥中报道过,该研究发现64份材料中只有37份材料足以恢复90% 的 NLR 基因数量。抗病基因通常以紧密连锁的物理簇组成,其中一些具有高度的变异性。成簇和非成簇抗病基因之间的差异可能是由于不等交换和减数分裂不稳定性造成的。
虽然可变基因倾向于富集在植物抗病上,但并非所有植物物种都是如此。例如,无油樟泛基因组的可变基因中含有的抗病基因就相对较少,而这些抗病基因却在核心基因中较多(胡海飞,个人交流) ,这一事实可能反映了该物种不同寻常的地理位置和进化历史。可变基因还常常与非生物胁迫和环境适应有关,这表明这些基因可能辅助未来的作物育种策略。
Origin of variable genes in plants
虽然对基因 PAV的鉴定已经非常常见,但对可变基因的发生机制了解相对较少。在植物中已经描述了几种可能导致基因获得或丢失的机制(图3)。新基因可通过全基因组复制、局部串联重复、 TE介导的复制、片段复制、近缘物种的融合、基因水平转移和从头产生等方式产生。基因也可能由于缺失而丢失,如由于染色体内重组和假基因突变导致的基因缺失。通过对植物泛基因组的分析,可以更全面地了解不同的基因获得和丢失机制对整个物种基因含量的相对贡献,并有助于我们理解选择导致变异基因频率发生变化的机制。
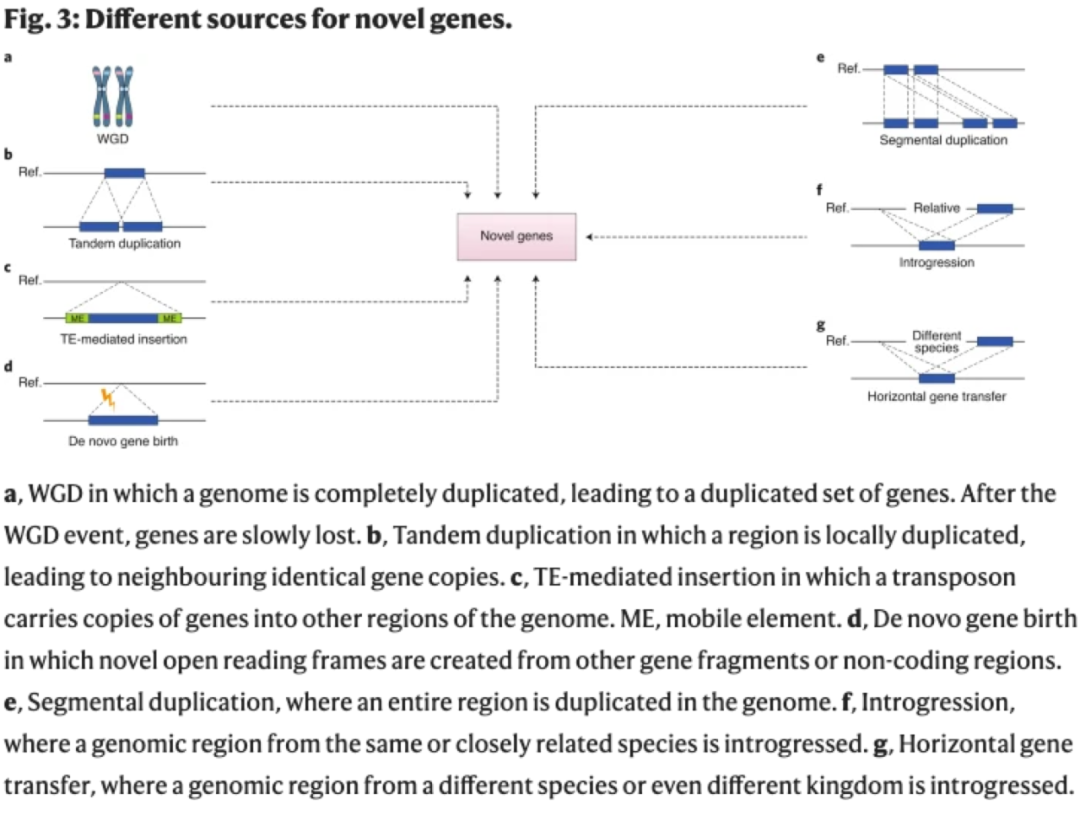
多倍体物种的可变基因比例似乎高于二倍体物种,然而目前可用于证实这一趋势的多倍体泛基因组很少。异源多倍体中可变基因的含量可以通过显性亚基因组形成,正如在草莓和油菜亚基因组之间观察到的那样,显性亚基因组承载着更大比例的核心基因。全基因组复制常常伴随着基因丢失。例如,芸薹属植物经历了一个全基因组三倍化(WGT)事件和随后的分化,产生了三个亚基因组(LF、MF1和MF2)。接下来的泛基因组分析揭示了亚基因组和可变基因比例之间的显著关联,LF 含有的可变基因最少,MF2含有的可变基因最多。可变基因在芸薹属的亚基因组位置可能与基因丢失率相关,即使在非常短的进化尺度上,也会影响种内变异。然而,大多数的油菜可变基因并没有这种亚基因组分布的规律,这与在短柄草泛基因组中观察到的现象类似,即可变基因所在的区域与其他禾本科同源区域的共线性较差,这表明它们更有可能位于共线性区段之外。芝麻在大约7000万年前经历了 基因组多倍化(WGD),这项泛基因组研究的目的是确定核心基因和可变基因的起源。几乎一半的核心基因和只有约10% 的可变基因可以追溯到 WGD。WGD 起源的可变基因所占比例较低,反映了许多基因不存在于共线性区段中。同样比例的核心基因和可变基因(约10%)可以归属于局部串联复制,这表明,对于芝麻来说,尽管品系特异性变异确实存在,但串联复制不是可变基因的主要来源。
部分同源交换(HE)是双多倍体植物基因 PAV 的另一个常见原因。在甘蓝型油菜中发现HE与表型变异有关。具有方向偏好性的HE可以导致亚基因组优势,这一现象也在其他物种中存在,包括多倍体草莓、小麦、咖啡和棉花等。对甘蓝型油菜全基因组的分析揭示了两种类型的基因 PAV 事件: non-HE-PAV,其中个体基因是可变的; HE-PAV,由于大片段的基因组交换引起的连续基因缺失。
植物泛基因组研究发现转座子(TEs)在基因多样性形成过程扮演着重要的角色。TE与基因 PAV、基因转移之间的关系已被广泛研究。最近的泛基因组研究使得 TE 对可变基因的影响有了更进一步的认识:物种内 TE 动态变化是导致基因产生和丢失的重要因素。甘蓝型油菜和短柄草中的可变抗病基因倾向于与 TE共定位。此外,基于8个个体全基因组组装的甘蓝型油菜全基因组分析,揭示了 TEs 在农艺性状中的作用。自从芭芭拉·麦克林托克在玉米中发现TE以来,TE 活性一直与基因组重组有关,从那时起,许多导致基因 PAV 的 TEs 被发现。未来的泛基因组研究将有助于我们理解 TE-PAV 相关性;例如,调查某些 TE 家族是否更有可能与 PAV 相关,以及这些关系是普遍存在的还是物种特有的。不断改进的TE预测和分类方法,将加深我们对 TE 在基因PAV变异方面的理解。
如何从头产生新基因还未被深入研究。最近对13个亲缘关系密切的普通野生稻基因组进行了比较分析,确定了175个从头产生的新基因。长非编码 RNA (lncRNA)在进化上要比蛋白质编码基因短,往往具有较高的组织表达特异性。在水稻中约91% 从头产生的新基因来自非编码转录本。因此,对 lncRNA 进行详细的注释和分析,可以扩展植物的可变基因库。
Prospects and future directions
随着长读序列数据质量的提高和以及成本的迅速下降,基因组序列数据日益增加,继而将促进泛基因组研究。未来单一参考基因组可能变得多余,而泛基因组日益成为新的参考,从而加深对基因组进化、驯化和功能等方面的了解。
泛基因组分析的挑战之一是泛基因组数据的存储和可视化。使用vg或 MGR工具可实现存储整个群体变异的泛基因组图。有必要建立基于泛基因组分析的基因组变异和注释分析。在植物育种种群中,可利用单体型图进行泛基因组构建。这些基因组图依赖于参考基因组坐标系和基因位置信息,使他们能够避免高度重复和高度分化区域。
对基因和基因组进行准确和一致的功能注释的方法远远落后于其组装的方法,尤其是许多可变基因的作用仍然是未知的。我们确实知道可变基因具有某些特点,如同源性较低,进化速度较快,表达水平较低等。对核心基因和可变基因之间的功能和相互作用的理解将极大地增加泛基因组研究的价值。一种可能的方法是采用整合基因组学的方法,如基因表达、共表达网络和序列保守性等与其功能联系起来,以便更广泛地了解其潜在功能。
迄今为止,大多数泛基因组研究都集中在基因组的基因部分,然而基因组以外的区域解释了作物表型变异的相当大的比例。许多重要的农艺性状可能是由基因调控的变化而不是基因 PAV 决定的。例如,在番茄泛基因组中发现的正在被选择的区域包括一个与风味相关的启动子。结合表观基因组数据对基因的调控区注释,泛基因组可以挖掘调控序列变异,继而在育种上利用。
近年来,原核生物的泛基因组已经跨越了种甚至种界,其中包括一项来自10个原核生物门的7,104个基因组的研究。由于原核生物单倍体基因组小,这种研究在计算上是可行的。然而,在植物中,目前还没有泛基因组跨越属的边界,可能是由于计算和经费的限制。随着测序成本继续下降,计算能力上升,植物泛基因组研究可能会超越物种层面,以至于我们可以开始在属乃至家族层面上构建泛基因组,从而使我们能够提出一些问题,比如什么基因是豆科植物必须的。最终,这将使我们能够预测和描述所有植物物种的基因集合,这些知识将彻底改变未来的基因组研究。如此广泛的全基因组将允许我们回答一个古老的问题: 是什么基因造就了植物?
原文链接:
https://www.nature.com/articles/s41477-020-0733-0
https://blog.sciencenet.cn/blog-1094241-1292306.html
上一篇:GATK流程不用再分割小麦染色体 part2
下一篇:翻译|三代测序在群体水平上的研究进展