博文
碱性水电解绿氢技术  精选
精选
|
全球能源格局正在发生重大变化。氢气被视为未来的能源载体,将成为发展更可持续的工业和社会的关键因素。然而,目前氢气主要由化石燃料生产,这种情况需要改变。采用先进的碱性水电解技术在利用可再生能源生产大规模绿色氢气的过渡中具有最大的潜力。工业电解槽设备的组装在更大范围内更为复杂,但它遵循一个基本的工作原理,即涉及阳极和阴极位点的两个半电池,其中发生析氧反应 (OER) 和析氢反应 (HER)。在这两种反应中,碱性水电解在热力学和动力学上都更具挑战性。除了获得可再生电力外,为碱性水电解开发耐用且丰富的电催化剂仍然是大规模碱性水电解的一个挑战。在不同的理化性质中,电催化剂表面及其与水和反应中间体的相互作用,以及形成的分子氢和氧,对催化性能和反应机理起着至关重要的作用。特别是,催化剂表面与中间体之间的结合强度决定了限速步骤和电催化性能。本文通过介绍氢经济的基本原理和碱性水电解的基本原理,对氢经济现状和碱性水电解的基本原理进行了一些深入介绍。此外,还简要讨论了碱性水电解的HER和OER反应机理以及两种半反应的电催化剂研究进展。本文通过具体参考文献解释了OER的吸附物演化机理和晶格氧机理。本综述还阐述了在开发过渡金属基电催化剂用于碱性水电解方面的选定贡献,特别强调了OER。重点关注了提高本征活性、电子填充的作用、相分离以及钴基电催化剂的缺陷结构对OER的影响。进一步讨论了钴氧化物电催化剂在工作条件下的结构改性和相变。此外,还讨论了通过从碱性电解质中吸收铁来创造新的活性表面物种以及激活钴基和镍基电催化剂的过程。最后,本综述简要概述了与大规模生产和利用绿色氢气相关的挑战。

Tüysüz H. Alkaline Water Electrolysis for Green Hydrogen Production. Acc CHem Res. 2024 Feb 9.
一、引言通过电解水生产氢气是将电能转化为化学能的主要选择之一。清洁氢被认为是未来的燃料,因为它可用于各种应用和部门,包括交通运输,以及家庭和工业的供热和发电。2022年全球 H2需求增长3%,达到9500万吨。几乎所有的H2工业使用燃料如天然气和煤炭制备。2022 年的制用氢过程产生了约 900 公吨的 CO2排放。由水分解产生的绿色H2量(只,占当今全球 H2 产量的 0.1%, 以化石燃料为基础的来源生产、碳捕获、利用和储存的蓝色 H2仅为0.7%左右。
最近的一项分析显示,蓝色 H2生产并不像人们认为的那样是一个低排放的过程。据计算蓝色 H2 的总二氧化碳排放仅比灰H2 低9-12%,灰氢是通过天然气的蒸汽重整生产的。经计算,蓝色 H2 的总体温室气体足迹比燃气或燃煤直接发热高出20%以上。使用可再生电力通过电解水生产合成H2将是收获零排放清洁氢用于未来的可持续应用的唯一选择。
截至2022年底,全球电解水H2装机容量产量达到近700兆瓦,与上年相比增长了约20%。如果所有计划中的项目都投入运营,到2030年,全球电解能力可能达到175吉瓦。如果包括早期项目,这一容量甚至可以增加到420吉瓦。虽然这是非常令人印象深刻的进展,但到 2030 年,要实现净零排放,将需要约 600 吉瓦电解能力的运营目标。因此,计划中和尝试的项目需要以更快的速度扩大和安装。
二、碱性水电解(AWE)2.1. 基本原则电化学水分解由两个半反应组成,即析氢反应(HER)和析氧反应(OER)。如图1a所示,这些反应分别发生在阴极和阳极位点,并且优选通过离子交换膜隔开。在其他参数和指标,如温度、压力、电解液的类型和浓度、电极类型和电池配置中,HER 和 OER 都对电解液的pH值非常敏感,并且它们以不同的反应途径进行。
在酸性介质中,水被氧化成分子氧,在阳极产生质子。生成的质子被转移到阴极并还原成分子氢。在碱性条件下,如图1a所示,由阴极位点的水产生的氢氧阴离子充当电化学电荷载流子。氢氧阴离子在阳极位点的氧化产生氧气分子和电子,进一步用于 H2在阴极现场产生。电化学反应发生在电极表面与电解质的界面处,根据法拉第电解定律,气体逸出速率与流经电路的电流成正比。尽管工业电解槽设备组件在更大的规模下更为复杂(图 1b),其通常由电解槽电池组 H2和 O2分离罐、冷却器和循环泵,其基本工作原理基于两个半电池。
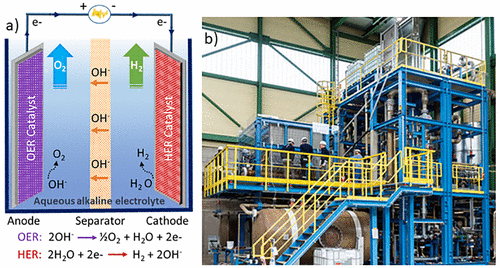
图 1.(a)基本AWE电池的图示,该电池由电路、电解质、隔膜以及阳极和阴极电极组成,阳极和阴极电极分别用活性催化剂装饰,其中分别发生OER和HER。(b)2兆瓦水电解厂。
在热力学上水分解是一个耗能反应,需要 ΔG = 237.1 kJ/mol 的能量输入。换言之,驱动水电解需要1.23 V的热力学电位。实际上,需要更高的外加电压,即所谓的过电位(外加电位与理论值1.23 V之间的差值)来克服几个障碍。这些障碍包括电路的电阻、电化学反应的活化能以及与气泡相关的障碍,这些障碍会导致对离子转移和电化学反应的额外阻力,以及质量传输,特别是由于OER缓慢。为了使该过程更经济,应降低两个半反应的过电位。这可以通过使用可以减少能量输入和活化能的电催化剂来实现,这将在第 3 节中讨论。
2.2. 工业碱性水电解槽的配置碱性水电解(AWE)是氯碱工业中用于大规模氢化的最实用、最先进的技术之一,具有悠久的应用历史。它通常在 50–80°C 的温度范围内工作,压力高达 30 bar。在经典的工业配置中,由雷尼镍、镀镍(或镀铁)钢或镍/不锈钢网制成的电催化剂负载电极浸入高浓度的 KOH 水溶液(通常为 20-30 wt %)中。由固体多孔氧化物制成的隔膜(如Zr基Zirfon Perl UTP 500,Agfa-Gevaert N.V.),允许运输OH–电极之间用于分离形成的H2和 O2气体。该分离器具有低气体污染和高离子电导率,其性能会根据几何结构和成分而波动。隔膜是电池整体性能的重要组成部分,因为它确保电极之间的离子接触并防止产生的气体混合。
使用两种不同类型的电解槽作为支架,即简单的罐式电池(单极)和由多个堆栈组成的压滤池(双极)。单极电池非常紧凑,欧姆损耗较低,而双极电池具有更复杂的结构设计,需要电解质循环和使用外部气体分离装置。电解槽也可以分为传统和零间隙组件。传统的电解槽组件基于在阳极和阴极电极之间保持一定的距离,而在零间隙电池的情况下,电极和隔膜以逐层配置压在一起,以最小化阳极和阴极位点之间的距离。在零间隙电解槽配置中,可以减少由于电路中电子的重新定位以及离子通过电解质和隔膜的运动而导致的压降相关的欧姆损耗。但是,这也带来了一些缺点。减小距离会导致两个电极之间的气泡积聚增加,并降低电解质的电导率。工业 AWE 的主要缺点之一是它必须在较低的电流密度下运行,通常在 0.05 至 0.7 A/cm2 的范围内,取决于由于多孔膜的不渗透性行为而产生的细胞压力,用于气体分离。由于对绿色H2的需求和兴趣不断增加,AWE开始受到更多关注。这种指数级的增长和兴趣迫使学术界和工业界重新审视和改进其某些方面,特别是开发低成本和稳定的电催化剂。(我简单理解是,碱性水电解本来已经非常成熟,但用于绿色氢气制造仍然存在许多需要改进的余地,特别是更好的催化剂,更好的鲁棒性和规模性要求)
三、碱性水电解电催化剂电催化剂的活性取决于材料的几个物理化学性质,包括成分、电导率、电子和晶体结构、形貌和结构参数,以及制备方法、晶界、表面结构和缺陷的存在。电催化剂的性能可以通过增加给定电极上的活性位点数量,通过增强负载或改变结构特性以暴露更多的催化活性位点来提高,或者通过保持质量恒定来操纵每个活性位点的内在活性。催化剂材料必须满足一些基本要求和标准,才能考虑用于大规模应用。一方面,它应该是高效的,在低施加电位下提供高电流密度,在工作条件下应具有良好的结构耐久性和稳定性,并且应该具有成本效益。另一方面,催化剂的品质因数应是整体的,并在电催化剂设计过程中考虑其他关键方面,包括可持续性、临界性(包括原材料的供应和地缘政治风险)、生态和可回收性。考虑到资源有限和许多元素在不久的将来枯竭,在生产的每个阶段优先考虑可持续性和可回收性非常重要。值得注意的是,催化剂的电催化性能在很大程度上取决于实验条件和测量技术。如需更详细地了解电催化性能和性能指标,以及评估OER催化剂活性和稳定性的方案,可以参考其他综述文章。
3.1. HER的电催化剂和反应机理催化剂表面及其与水和反应中间体的相互作用,以及形成的分子氢,在催化性能和HER机理中起着至关重要的作用。在碱性介质中,催化剂上的双电子转移HER机制是基于水的O-H键的裂解,电极表面氢物质的吸附,H-H键的形成,以及最终分子H2的释放。图 2a 所示,HER 从 Volmer 步长开始,其中 H2O被还原,生成的原子氢键合到电催化剂的表面(以H*形式提供)。随后与另一个氢原子结合并解吸 H2,这可能通过 Tafel 反应(两种吸附氢的组合,H*)或 Heyrovsky 途径(另一种氢参与 H2O分子的解离)发生,如图2所示。
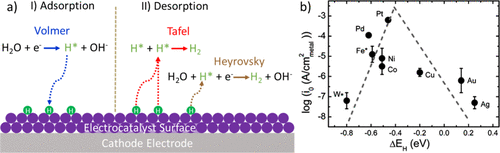
图2.(a)由吸附(Volmer)和解吸(Tafel或Heyrovsky)步骤组成的碱性HER的示意图和(b)典型HER电催化剂在碱性介质中活性的火山形曲线,其中交换电流密度(log(i0)) 在单金属表面上绘制为计算出的氢结合能的函数。
氢吸附/解吸步骤和金属-氢(M–H)键的强度决定了 HER 动力学。根据萨巴蒂尔原理,M-H键既不能太强也不能太弱,以形成反应中间体和分子氢。氢结合能是电催化剂活性的一个非常重要的指标,它被普遍认为是最终的性能描述符。交换电流作为 M-H 键强度的函数,显示出火山形曲线,Trasatti 在 1970 年代初期使用 H2 的实验数据对其进行了描述在酸性介质中进化。Nørskov等人利用密度泛函理论(DFT)计算的电化学反应中间体的吸附能作为电极电位的函数,建立了现代火山图。如图2b中的火山形曲线所示,Pt电催化剂在碱性电解质中表现出最高的电流密度和优化的氢结合能。这与Trasatti描述的酸性介质中的情况相当。
虽然这种相关性对预测活性非常有利,但实际上,氢吸附能与电催化剂性能之间的相关性对于HER更为复杂。大多数金属(尤其是过渡金属)在碱性电解液中易被氧化,其表面被氧化层覆盖。因此,氢原子不会与金属直接接触。这导致催化剂在火山图中的位置发生变化。此外,反应动力学也会受到其他物质(如OH–),可以阻断活性位点并改变吸附氢物种的能量状态。
铂族金属是众所周知的HER电催化剂,其中Pt通常用作碱性和酸性介质的基准电催化剂。然而,Pt的高成本和有限的资源需要寻找替代的电催化剂。镍基电极也被广泛用作 H2 的阴极工业AWE中的发电。然而,原始的镍电极在恶劣的电解条件下会发生结构变化。这导致电极表面的改变和其他物质(如氢化镍)的形成,从而导致快速失活。由镍-铝制成的雷尼镍合金已被用于替代HER催化剂,具有更高的稳定性。据报道,在一系列镍基二元合金中,通过电沉积在钢带上生产的Ni-Mo合金显示出非常好的活性,在6 M KOH中连续电解超过1500小时,在300 mA/cm2时仅具有约180 mV的过电位。
纳米Ni(OH)2的沉积还发现Pt电极表面的簇使HER活性增加了8倍,从而Ni(OH)2的边缘团簇可以促进水的解离和氢中间体的生成。对具有Ni-Fe和Ni-Co氢氧化物团簇的Pt(111)单晶的表面装饰进行了类似的观察(图3a,b)。这些团簇促进了水的解离,以及在0.1 M KOH电解液中达到10 mA/cm2电流密度所需的过电位降低至约70 mV。Fe的添加是为了帮助Ni团簇解离水,Ni-Fe团簇增加的*OH结合能支持了该提案。据报道,负载在碳纳米管上的氧化镍/镍异质结构具有与商业Pt/C催化剂一样好的HER活性。催化剂的高性能归因于异质结构中的NiO/Ni界面,其中NiO和Ni位点可以作为OH–的吸附位点和氢气。对于嵌入在N掺杂碳上的钴-钴氧化物杂化物,也可以观察到类似的协同作用,N掺杂碳可以用作AWE的阴极和阳极材料。有关AWE的HER电催化剂开发的更全面的回顾,请参见Lim最近发表的综述文章。
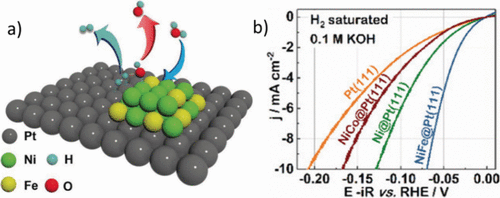
图3.(a) Ni 和 Fe 金属氢氧化物团簇修饰的 Pt(111) 电极上 HER 的机理。提出了Ni-Fe团簇来改善初始水解离步骤(蓝色箭头),Pt刺激氢物种和H的吸附2气体逸出(玉箭)。(b) 在0.1 M KOH中沉积在Pt(111)单晶上的金属氢氧化物团簇的极化曲线。
3.2. 碱性水电解的电催化剂和反应机理
OER机理更为复杂,因为它具有几种中间状态,具有不同的活化步骤,这些步骤会影响其反应速率。吸附物演化机制 (AEM) 是最广泛接受的机制。如图4a所示,OER在碱性介质中的第一步是吸附氢氧根离子(OH–)在电催化剂的活性位点(表示为M),M-OH中间体的形成和电子的释放。M-OH中间体与另一种OH–的进一步反应离子导致 M-O 中间体和 H2O 的形成,以及另一个电子的释放。分子 O2可以通过两种不同的途径从M-O中间体形成:(i)两种M-O物种的直接结合;(ii) OH–离子的亲核攻击M-O,生成 M-OOH 中间体及其与另一种 OH–离子的进一步反应,最后转化为水和分子 O2。第二种途径是最普遍接受的 4e–在碱性介质中的转移反应机理。
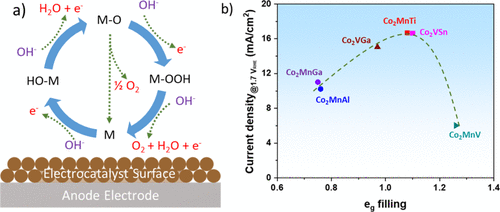
图4.(a) 催化剂表面的 OER 机理草图和 (b) 钴基 Heusler 化合物的 OER 活性的火山形曲线,1.7 V 时电流密度与 RHE相对钴的eg电子的占有率。
催化剂表面与OH–的结合强度离子、中间体(HO*、O* 和 HOO*)和产物决定了电催化剂的限速步骤和整体 OER 效率。早在1955年,人们就已经认识到碱性介质中OER的M-OH键能变化与所需过电压之间的相关性。如果催化剂表面与氧的结合力太强,则反应速率会受到 HOO* 物质形成的限制。另一方面,如果催化剂表面与氧的结合太弱,则电位会受到HO*中间体氧化的限制。将电催化剂的性能绘制为结合能的函数,可得出火山形曲线,其中氧化物如 Co3O4和 RuO2和钙钛矿,如LaNiO3和 SrCoO3由于其最佳结合能,显示出最低的理论过电位。这与RuO电催化活性序列的实验数据有很好的相关性2>IrO2> 含 Co 和 Ni 的氧化物 >含 Fe、Mn 和 Pb 的氧化物。尽管如此,理论预测与实验测量的金属表面OER活性之间的可靠相关性并不简单,因为反应发生在碱性介质中的氧化表面上。
作为AEM的替代方案,最初在LaNiO3上提出了晶格氧机制(LOM)电催化剂,被发现具有较低的反应势垒。AEM和LOM中的OER过程都从催化剂表面的羟基化开始。然而,LOM涉及氧化中间体和催化剂晶格氧的直接O-O偶联。尽管反应中间体相似,但LOM的不同之处在于晶格氧演化过程中氧空位的产生,这与特定质子-电子转移步骤的解耦有关。这导致了pH依赖性的 OER 动力学。晶格氧在钙钛矿催化剂内OER机理中的参与也已通过使用电化学质谱测量得到验证18O标记的钙钛矿。已经表明,通过取代催化失活的Zn2+,OER机制可以从AEM切换到LOM进入CoOOH 结构。Zn2+的添加发现离子会产生具有不同局部构型的氧非键态。
许多物理化学性质会影响材料的OER性能。Markovic 等人证明 OH–的强度OHad–Mn+和3d金属氢氧化物催化剂中的相互作用主要决定了催化活性。电子结构,特别是金属d波段中心相对于其费米能级的位置,在OER中间体和分子O2中起着重要作用吸附强度。作为吸附剂(OH–离子)接近催化剂表面,吸附物的电子与金属的价S、P和D带相互作用,并通过金属-吸附物键形成中间体。金属-吸附物键的稳定性和反应性很大程度上取决于 d 电子的数量。SHao-Horn等人进一步发现,–eg表面过渡金属阳离子的填充会影响OER中间体与氧化物表面的结合,从而影响OER性能。本文作者的团队最近证明,类似的碱性水电解活动,g还可以观察到钴基Heusler化合物(Co2YZ)在碱性电解质中。如图 4b 所示,eg催化活性Co位点的轨道填充可以通过改变的Y位点和Z位点来调节Co2YZ化合物,其中与接近统一eg轨道填充的Co2MnTi 和 Co2VSn化合物的催化性能更高。理论预测支持通过两种 M-O 物种的直接组合对 Heusler 化合物进行更优选的 OER 途径。eg轨道填充的改变可以调节催化剂的M-O键结强度和OER活性。
氧化物电极,称为尺寸稳定阳极 (DSA),具有成型的电化学技术。1970 年代初和 1980 年代,Trasatti报告了 RuO2和 IrO2电极的卓越的析氢和析氧性能。IrO2曾经被用作最先进的OER电催化剂。然而,由于其成本高且资源有限,人们投入了大量精力来寻找替代的OER催化剂,特别是基于丰富的第一排过渡金属,如Mn、Fe、Co和Ni。下一节将详细介绍有关镍基、铁基和钴基氧化物的部分报告,包括作者实验室的研究,重点是改善电催化剂的本征特性和铁杂质在碱性电解质中的作用。
Corrigan在1987年的研究表明,Ni和Fe之间对OER活性具有明显的协同作用。从商业KOH电解质中引入的Fe杂质或在薄的氧化镍电极中共沉淀的Fe杂质对电催化剂的性能有很强的影响。BoettcHer等人揭示了Ni(OH)2/NiOOH 的电导率在与 Fe 共沉淀时至少增加 30 倍,这会产生对 Ni 的部分电荷转移活化效应,从而导致 OER 增强,类似于在贵金属电极表面观察到的。混合Ni-Fe氢氧化物(Ni1-xFexOOH)在其纯Ni和Fe母体化合物上也与异常短的Fe-O键距离有关,这是由与[NiO]6周围的边缘共享引起的八面体,这导致 OER 中间体的最佳吸附能和 Fe 位点的低过电位。作者团队的一项系统研究揭示了成分的明显影响以及NiFe氧化物中Ni/Fe比对OER的作用。以茶叶孔隙限制为模板,可以制备一系列平均粒径约为8 nm、织构参数相似的Ni-Fe氧化物纳米颗粒。由于KOH电解液中Fe杂质的吸收,观察到所有含Ni样品在电化学循环过程中都经历了活化过程。
尽管结晶氧化钴尖晶石在 KOH 电解质中电化学循环时失活,但它与镍的结合已被证明可以提供从 KOH 电极吸收铁的能力并提高 OER 活性。当银部分(金属Ag和氧化银)与结晶钴氧化物结构偶联时,同样可以实现这种活化(图5a,b)。作者的团队证实,通过纳米铸造路线将银与有序的介孔钴氧化物结构相结合对其OER活性具有双重影响。发现金属Ag的掺入增强了氧化物的电导率,而氧化银物种导致了Fe诱导的电催化剂活化。如图 5c 所示,虽然原始 Co3O4经过50次循环伏安(CV)实验后,所有含银样品均被激活,所需电位达到10 mA/cm2滴入含有铁杂质的1M KOH中。
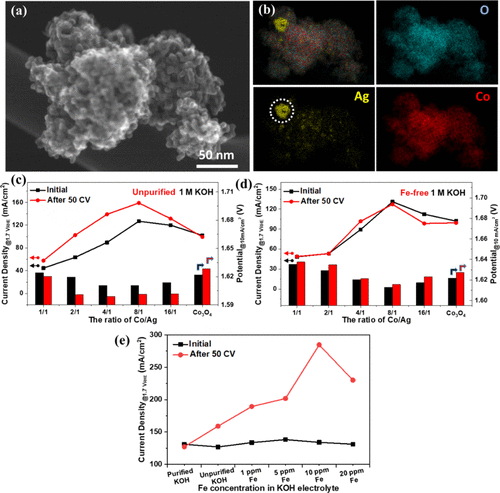
图5. SEM银-钴复合氧化物图像(a)选定的银-钴复合氧化物相应元素映射图像(b)。1.7 V 时的电流密度热和 10 mA/cm2 时的施加电位原始公司 50 CV 之前和之后Co3O4和 1 M KOH 电解质中的 Ag–Co 氧化物,纯化后含有痕量 Fe 杂质 (c) 和不含铁的 1 M KOH (d)。1.7 V 时的电流密度热选定的 Co8Ag纯化的 1 M KOH 中的银氧化物,具有不同的 Fe 浓度,并控制添加 Fe(e)。
当商业KOH纯化并用作电解质时,未观察到活化(图5d)。当在纯化的KOH电解液中加入控制量的铁时,选择最活跃的钴-氧化银电催化剂再次进行活化过程,而10 ppm铁杂质的存在导致电流密度最高,在1.7 V 时几乎达到300 mA/cm²,如图 5e 所示。
与结晶相比,我们已经验证了通过电化学沉积法制备的无定形氢氧化钴也能够从KOH电解质中吸收铁。总体而言,吸铁能力对于催化剂的再生和创建更活跃的催化中心来说是一个明显的优势。然而,其他杂质,如高浓度碱性电解质中的碳酸盐和有机物质,也可以沉积在Co3O4上,影响其表面结构和OER性能。
除了原位沉积之外,铁在合成氧化钴纳米线过程中的掺入通过增加钴中心周围的平均畸变以及在四面体和八面体位置上的Co2+与Co3+的比例,改变了它们的电子结构。此外,优化复合纳米线结构的还原导致了相分离,并形成了如图6a,b所示的氧化铁纳米颗粒装饰的CoO纳米线阵列。Co3O4尖晶石结构还原为CoO岩盐结构并形成相界,这有利于电荷转移。这导致在1.7 V vs RHE(可逆氢电极)下电流密度从150显著增加到315 mA/cm²(见图6c)。原位电化学拉曼光谱(图6d)和后结构分析表明,还原后的材料作为前催化剂,在氧气析出反应(OER)过程中转化为羟基氧化物物种和无序的氧化钴尖晶石。

图6.(a,b)还原的氧化铁纳米颗粒负载的CoO纳米线的TEM和HR-TEM图像,(c)Co/Fe比分别为32和3(分别表示为Co/Fe 32-红和Co/Fe 3-红)的还原CoO和CoFe氧化物的LSV曲线,原始Co3O4和 Fe3O4在1M KOH电解质中。(d)Co/Fe 32-红样品的原位电化学拉曼光谱,其中CoO转化为Co3O4在更高的应用电位下可以进行监测。
作者的团队还证实,将硼与氧化钴结合可以改变其结构并提高碱性水电解的性能。采用简易沉淀法制备了一系列形貌、结晶度和结构参数各不相同的Co-B氧化物4作为还原剂和B源。在高温下处理成品材料诱导结晶Co3O4的形成和无定形氧化硼。硼的存在影响了形貌、结晶和表面结构,导致OER活性增加3倍。在不同的复合氧化物中,在300 °C下煅烧的部分结晶样品通过提供235 mA /cm²的电流密度,对OER表现出最高的催化性能1.7 V 时热并且需要 338 mV 的过电位才能达到 10 mA /cm².电催化剂的改变和新物质的形成(如CoO2和(氧)氢氧化物)在OER期间被发现高度依赖于样品的结晶度。这一发现证明了新组合物在提高能源效率和推动该领域发展方面的潜力。
综上所述,除实验条件外,催化剂的各种理化性质,如形貌、尺寸、尺寸、形状、组成、晶体和电子结构、表面积、掺杂、缺陷,以及催化剂和电极的制备方法,都会显著影响其OER活性。监测结构改变并捕获表面中间体和活性位点将揭开水电催化的奥秘,从而更好地了解该过程并开发更具活性、更耐用和最实惠的催化剂。这将是未来大规模绿色H2的绝对游戏规则改变者生产以满足日益增长的需求。
四、总结与展望碱性水电解是绿色制氢的成熟技术,在规模化生产中越来越受到关注。然而,仍然需要优化工艺并开发更实惠、活性和耐用的电催化剂,特别是对于要求更高的碱性水电解。对于催化剂的开发,必须更好地了解工艺和反应机理。原子经济性是可持续催化剂设计应考虑的一个关键生态和经济点。也可以考虑一步制备电催化剂工作电极,以降低制造成本。
商业KOH中的铁杂质可以激活一些电催化剂,例如无定形氢氧化钴和镍基氧化物以及用于OER的含镍、含银钴氧化物。虽然这在小规模的实验室规模上可能是有利的,但对于使用高浓度KOH水溶液作为电解质的工业应用来说,它可能会有问题。工业级和试剂级KOH的纯度分别约为85%和90%,并含有其他物质,如氯化物、碳酸盐、磷酸盐、硫酸盐和甲酸盐和乙酸盐等有机物,以及铁和铅等金属。这些污染物的大量沉积物,尤其是碳酸盐和铁,会阻塞催化位点并导致催化剂失活。在AWE试验工厂中研究KOH中杂质的影响是值得的。
绿色氢气正在改变我们的能源格局,并将在社会和可持续未来中发挥重要作用。绿色氢的重要性已得到公认。绿色氢正在被迅速推动,以取代能源载体并升级许多工业流程。然而,既没有足够的产能来生产大量的绿色氢气,也没有合适的基础设施来利用绿色氢气。绿色氢气生产的成本和可及性主要取决于可持续电力的价格及其可用性。此外,可再生能源的波动性是碱性水电解厂的另一个限制因素,这些电解厂需要固定的运行条件。作为一个社会,我们仍然没有达到可以从可再生能源中生产常规电力的地步。因此,应该在可持续电力生产方面进行更多投资,特别是制造陆上和海上风力涡轮机和光伏太阳能电池板。可持续电力将是大规模和廉价制氢的最大瓶颈。人们相信,非洲丰富的太阳能和风能可以使该地区成为成功的绿色氢生产国和全球出口国。然而,大约40%和60%的非洲人口分别无法获得足够的电力和生活用水。非洲大陆的资源是否应该出口,还是应该用于非洲大陆的发展,这是非常值得探讨的。
在绿色氢气生产之后,其储存、运输和安全监管将为广泛实施带来额外的挑战和问题。储存大量氢气,无论是气体还是液体,在技术和实践上都具有挑战性。作为气体,它需要非常高的压力罐,而作为液体,由于其沸点约为 20 K,它需要低温。 氢气也可以储存在固体中,也可以以化学方式储存在分子中,例如氨。与其他储氢技术相比,氨的合成和分配已经很成熟。然而,氨分解是一个非常耗能的过程,需要额外的催化系统来产生氢气。目前的技术也无法避免气态NOx的形成,高纯度制氢仍然具有挑战性。需要工艺优化和低温氨裂解催化剂的开发。
此外,值得注意的是,经济和基础设施方面的挑战也可能成为广泛采用绿色H2的障碍在工业应用中。例如,让我们考虑一家钢铁制造公司,该公司希望从使用煤炭作为能源转向使用绿色氢气。这种转变将需要大量的资本和投资来升级必要的基础设施。幸运的是,一些国家,如德国,为基础设施升级提供财政支持,以实现向更可持续的未来平稳过渡。
总体而言,不可否认的是,绿色氢气将在满足我们未来的能源需求方面发挥关键作用。然而,要实现大规模生产、储存和分配,以及与现有能源基础设施和其他部门(如工业、运输和发电)的整合,必须克服重大挑战。然而,氢气是通过能源转型为我们的社会实现可持续未来的关键。
原文作者信息Harun Tüysüz -马克斯-普朗克研究所多相催化和可持续能源部
https://blog.sciencenet.cn/blog-41174-1421379.html
上一篇:千年孤独
下一篇:血管周围成纤维细胞可能是ED的基础
