We first calculated the electron localization functions (ELF), known to be an informative tool to distinguish
different bonding interactions in solid17.
The isosurface plots at ELF = 0.75 (a typicalgood number for characterization of covalent bondings)17 clearly illustrate
the covalent bonding nature of the I-42d structure (Fig. 1c) and
confirm the formation of OH and H3O units in P21 structure (Fig. 1d; Supplementary Fig. S2).
We subequently performed a topological analysis of the static electrondensity through
Bader’s quantum theory of atoms-in-molecules18, which has beensuccessfully applied to
the determination of bonding interactions through
the values of the density and its Laplacian at bond critical points.
L is the bond length. ρ (rCP) and ∇2ρ(rCP) are the charge density and its Laplacian at the corresponding critical points, respectively.
The high charge densities and negative Laplacians ofcharge densities indicate a covalent bond, otherwise non-covalently bonded.
Densities and negative Laplaciansof increasing magnitudes indicate covalent bonds of increasing strength.The bold values indicate the predominant covalent bonds locatingin either OH or H3Ounit. The unit is in a.u.
Richard Bader from McMaster University(麦克马斯特大学,加拿大的一所著名大學),
developed an intuitive way of dividing molecules into atoms called the Quantum Theory of Atoms in Molecules (QTAIM).
His definition of an atom is based purely on the electronic charge density. Bader uses what are called zero flux surfaces to divide atoms.
A zero flux surface is a 2-D surface on which the charge density is a minimum perpendicular to the surface.
Typically in molecular systems, the charge density reaches a minimum between atoms and this is a natural place to separate atoms from each other.
Bader's theory of atoms in molecules is often useful for charge analysis.
For example, the charge enclosed within the Bader volume is a good approximation to the total electronic charge of an atom.
The charge distribution can be used to determine multipole moments of interacting atoms or molecules. Bader's analysis has also been used to define the hardness of atoms, which can be used to quantify the cost of removing charge from an atom. The theory also provides a definition for chemical bonding that gives numerical values for bond strength.
Grid Method
We have developed computational method for partitioning a charge density grid into Bader volumes which is efficient, robust, and scales linearly with the number of grid points.
The partitioning algorithm follows steepest ascent paths along the charge density gradient from grid point to grid point until a charge density maximum is reached. As the algorithm assigns grid points to charge density maxima, subsequent paths are terminated when they reach previously assigned grid points. It is this grid based approach which gives the algorithm its efficiency, and allows for the analysis of the large grids generated from plane wave based density functional theory calculations.
Code
The program, code, and information about running the program can be found here. <!-- The bader program can also be used to calculate site-projected DOS on Bader volumes. Details can be found here.
--> We have a discussion forum to address issues related to the code and running the program.
The Quantum Theory of Atoms in Molecules (QTAIM) is a model of molecular and condensed matter electronic systems (such as crystals) in which the principal objects of molecular structure - atoms and bonds - are natural expressions of a system's observable electron density distribution function. An electron density distribution of a molecule is a probability distribution that describes the average manner in which the electronic charge is distributed throughout real space in the attractive field exerted by the nuclei.
According to QTAIM, molecular structure is revealed by the stationary points of the electron density together with the gradient paths of the electron density that originate and terminate at these points. QTAIM was primarily developed by Professor Richard Bader and his research group at McMaster University over the course of decades, beginning with analyses of theoretically calculated electron densities of simple molecules in the early 1960s and culminating with analyses of both theoretically and experimentally measured electron densities of crystals in the 90s.
The development of QTAIM was driven by the assumption that, since the concepts of atoms and bonds have been and continue to be so ubiquitously useful in interpreting, classifying, predicting and communicating chemistry, they should have a well-defined physical basis.
QTAIM recovers the central operational concepts of the molecular structure hypothesis, that of a functional grouping of atoms with an additive and characteristic set of properties, together with a definition of the bonds that link the atoms and impart the structure. QTAIM defines chemical bonding and structure of a chemical system based on the topology of the electron density. In addition to bonding, AIM allows the calculation of certain physical properties on a per-atom basis, by dividing space up into atomic volumes containing exactly one nucleus, which acts as a local attractor of the electron density. In QTAIM an atom is defined as a proper open system, i.e. a system that can share energy and electron density, which is localized in the 3D space. The mathematical study of these features is usually referred in the literature as charge density topology.
QTAIM rests on the fact that the dominant topological property of the vast majority of electron density distributions is the presence of strong maxima that occur exclusively at the nuclei, certain pairs of which are linked together by ridges of electron density. In terms of an electron density distribution's gradient vector field, this corresponds to a complete, non-overlapping partitioning of a molecule into three-dimensional basins (atoms) that are linked together by shared two-dimensional separatrices (interatomic surfaces). Within each interatomic surface, the electron density is a maximum at the corresponding internuclear saddle point, which also lies at the minimum of the ridge between corresponding pair of nuclei, the ridge being defined by the pair of gradient trajectories (bond path) originating at the saddle point and terminating at the nuclei. Because QTAIM atoms are always bounded by surfaces having zero flux in the gradient vector field of the electron density, they have some unique quantum mechanical properties compared to other subsystem definitions, including unique electronic kinetic energy, the satisfaction of an electronic virial theorem analogous to the molecular electronic virial theorem and some interesting variational properties.
QTAIM has gradually become a method for addressing possible questions regarding chemical systems, in a variety of situations hardly handled before by any other model or theory in Chemistry[1][2][3][4]
QTAIM is applied to the description of certain organic crystals with unusually short distances between neighboring molecules as observed by X-ray diffraction. For example in the crystal structure of molecular chlorine the experimental Cl...Cl distance between two molecules is 327 picometres which is less than the sum of the van der Waals radii of 350 picometres. In one QTAIM result 12 bond paths start from each chlorine atom to other chlorine atoms including the other chlorine atom in the molecule. The theory also aims to explain the metallic properties of metallic hydrogen in much the same way.
The theory is also applied to so-called hydrogen-hydrogen bonds[5] as they occur in molecules such as phenanthrene and chrysene. In these compounds the distance between two ortho hydrogen atoms again is shorter than their van der Waals radii and according to in silico experiments based on this theory, a bond path is identified between them. Both hydrogen atoms have identical electron density and are closed shell and therefore they are very different from the so-called dihydrogen bonds which are postulated for compounds such as (CH3)2NHBH3 and also different from so-called agostic interactions.
In mainstream chemistry close proximity of two nonbonding atoms leads to destabilizing steric repulsion but in QTAIM the observed hydrogen hydrogen interactions are in fact stabilizing. It is well known that both kinked phenanthrene and chrysene are around 6 kcal/mol (25 kJ/mol) more stable than their linear isomersanthracene and tetracene. One traditional explanation is given by Clar's rule. QTAIM shows that a calculated stabilization for phenanthrene by 8 kcal/mol (33 kJ/mol) is the result of destabilization of the compound by 8 kcal/mol (33 kJ/mol) originating from electron transfer from carbon to hydrogen, offset by 12.1 kcal (51 kJ/mol) of stabilization due to a H..H bond path. The electron density at the critical point between the two hydrogen atoms is low, 0.012 e for phenanthrene. Another property of the bond path is its curvature.
Another molecule studied in QTAIM is biphenyl. Its two phenyl rings are oriented at a 38° angle with respect to each other with the planar molecular geometry (encountered in a rotation around the central C-C bond) destabilized by 2.1 kcal/mol (8.8 kJ/mol) and the perpendicular one destabilized by 2.5 kcal/mol (10.5 kJ/mol). The classic explanations for this rotation barrier are steric repulsion between the ortho-hydrogen atoms (planar) and breaking of delocalization of pi density over both rings (perpendicular).
In QTAIM the energy increase on decreasing the dihedral angle from 38° to 0° is a summation of several factors. Destabilizing factors are the increase in bond length between the connecting carbon atoms (because they have to accommodate the approaching hydrogen atoms) and transfer of electronic charge from carbon to hydrogen. Stabilizing factors are increased delocalization of pi-electrons from one ring to the other and the one that tips the balance is a hydrogen - hydrogen bond between the ortho hydrogens.
The hydrogen bond is not without its critics. According to one the relative stability of phenanthrene compared to its isomers can be adequately explained by comparing resonance stabilizations.[6] Another critic [7] argues that the stability of phenanthrene can be attributed to more effective pi-pi overlap in the central double bond; the existence of bond paths are not questioned but the stabilizing energy derived from it is
Jump up ^Bader, R. (1991). "A quantum theory of molecular structure and its applications". Chemical Reviews91: 893¨C928. doi:10.1021/cr00005a013.
Jump up ^Bader, R.F.W. (2005). "The Quantum Mechanical Basis for Conceptual Chemistry". Monatshefte fur Chemie136: 819¨C854.
Jump up ^Bader, R.F.W. (1998). "Atoms in Molecules". Encyclopedia of Computational Chemistry1: 64–86.
Jump up ^Hydrogen - Hydrogen Bonding: A Stabilizing Interaction in Molecules and Crystals Cherif F. Matta, Jesus Hernandez-Trujillo, Ting-Hua Tang, Richard F. W. Bader Chem. Eur. J. 2003, 9, 1940 ± 1951 doi:10.1002/chem.200204626
Jump up ^Polycyclic Benzenoids: Why Kinked is More Stable than Straight Jordi Poater, Ruud Visser, Miquel Sola, F. Matthias Bickelhaupt J. Org. Chem.2007, 72, 1134-1142 doi:10.1021/jo061637p
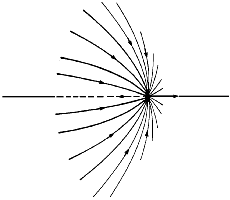
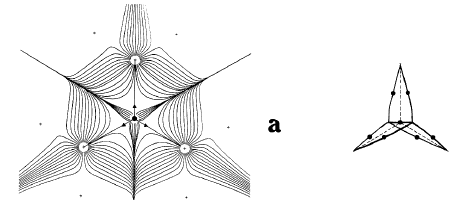
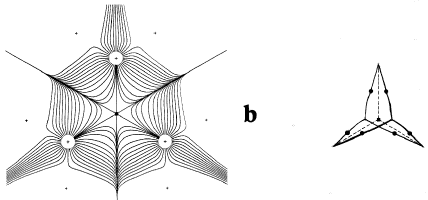
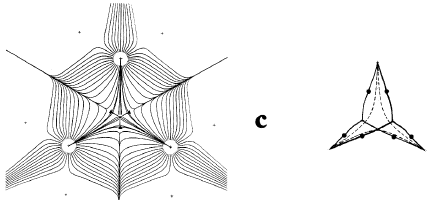
The second gradient vector field map in Figure 4 includes the trajectories, shown in bold, that both originate and terminate at the critical points found between nuclei that appear linked by a saddle in (r) in Figure 1. A critical point denotes an extremum in (r), a point where (r) = 0. Associated with each such critical point is a set of trajectories that start at infinity and terminate at the critical point, only two of which appear in the symmetry plane shown in the figure. They define an interatomic surface, a surface that separates the basins of neighbouring atoms. There is a unique pair of trajectories that originate at each such critical point and terminate, one each, at the neighbouring nuclei. They define a line through space along which the electron density is a maximum. The two sets of trajectories associated with such a critical point, a bond critical point, the set that terminates at the critical point and defines the interatomic surface and the pair that originates there and defines the line of maximum density, are shown in Figure 5.
Figure 5. A three-dimensional display of the set of trajectories of (r) that terminate at a bond critical point and define an interatomic surface and of the unique pair of trajectories that originate at the same point and define the bond path. Only one pair of each set that terminates at the critical point appears in the plane illustrated in Figure 4 (b) and (c).
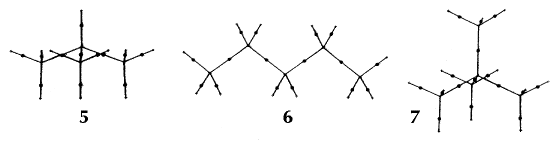
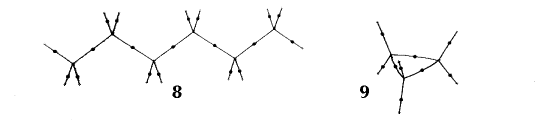
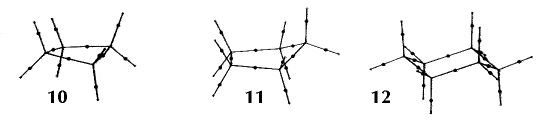
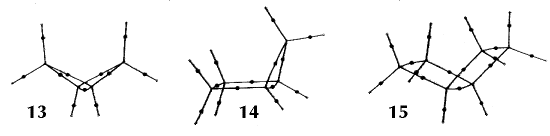
In an equilibrium geometry the line of maximum density is called a bond path because the set of bond paths for a given molecule, the molecular graph, faithfully recovers the network of chemical bonds that are assigned on the basis of chemical considerations. Thus a pair of bonded atoms are linked by a line along which the electron density, the glue of chemistry, is maximally concentrated. Molecular structures predicted by the molecular graphs determined by the electron density are shown in Figure 6.
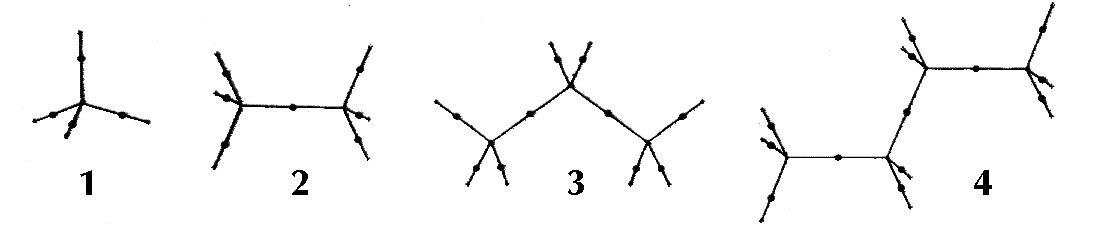
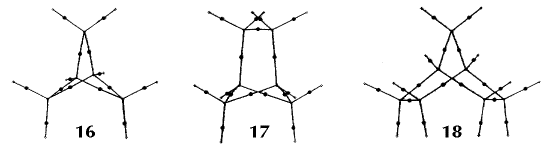
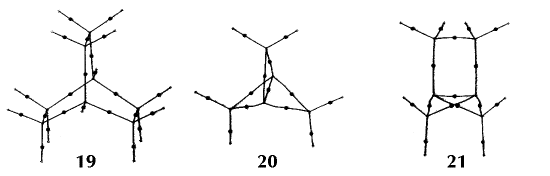
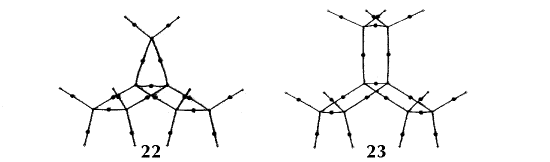
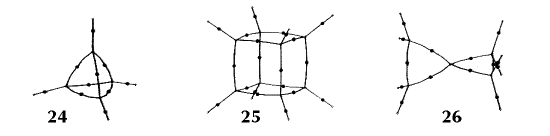
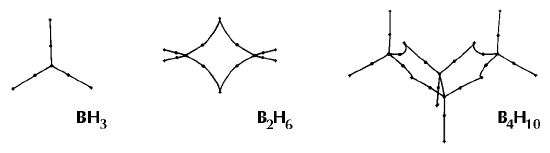
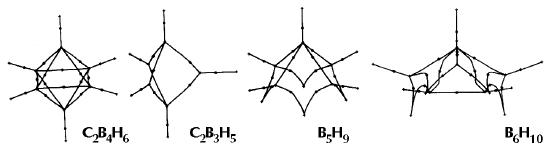
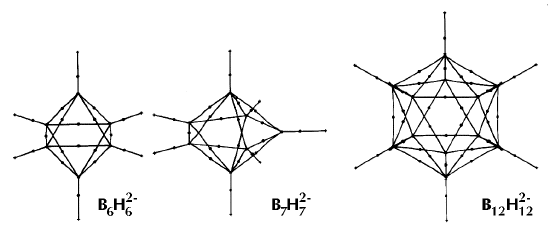
Figure 6. Molecular graphs - lines of maximum electron density linking bonded nuclei - in hydrocarbon molecules in diagrams 1 through 26, and boranes and carboranes below these. Bond critical points, where the trajectories defining the bond path originate, are denoted by dots. Note that the bond paths can be curved away from the internuclear axis in strained or in electron deficient molecules. A molecular graph and the characteristics of the density at the bond critical points provide a concise summary of the bonding within a molecule or crystal.
The molecular graph undergoes discontinuous and abrupt changes if the nuclei are displaced into critical configurations. When this occurs, one makes or breaks certain of the bonds and changes one structure into another. These changes are described and predicted using the mathematics of qualitative dynamics and the resulting theory of structural stability is illustrated in Figure 7 for the very strained molecule called [1,1,1]propellane.
Figure 7. Diagrams illustrating changes in structure induced by the dynamics of the nuclei. The molecular graph in a is for the highly strained [1,1,1]propellane molecule, C5H6 (the two hydrogens attached to each apical carbon atom are not indicated). The gradient vector field maps are for the symmetry plane containing the C-C bridgehead bond critical point and the three apical carbon atoms. When the separation between the two bridgehead nuclei is increased to a critical value, the bond critical point coalesces with the three neighbouring ring critical points to form a singularity in (r), as depicted by the gradient vector field map in b. The singularity is unstable and its formation signifies the breaking of the C-C bridgehead bond. Further separation of the nuclei causes it to bifurcate into a cage critical point yielding a new structure in which the bridgehead carbon atoms are not bonded to one another, the cage structure depicted in c.
分子中的原子理论(Atoms in molecules,简称AIM)是量子化学的一个模型。它基于电子密度(英语:electron density)标量场的拓扑性质来描述分子中的成键。除了成键性质之外,AIM 还根据拓扑性质对全空间进行划分,每个区域内正好包含一个原子核,这种区域给出了量子化学上定义原子的一种方式。通过对每一区域内进行积分,可以得到单个原子的一系列性质。AIM 方法于上世纪60年代由理查德·贝德(英语:Richard Bader)提出。在过去的几十年里,AIM 逐渐发展成一种用于解决化学体系中的许多问题的理论,其应用的广泛性远非之前提出的各种模型或理论所能及。[1][2]在 AIM 中,原子表现电子密度梯度场中的吸引子,因而可以通过梯度场的局域曲率来进行定义。这种分析方法一般在文献中称为对电子密度的拓扑分析,尽管这个词与数学中的拓扑一词的含义并不相同。
 Richard Bader from McMaster University(麦克马斯特大学,加拿大的一所著名大學),
Richard Bader from McMaster University(麦克马斯特大学,加拿大的一所著名大學),  Bader's theory of atoms in molecules is often useful for charge analysis.
Bader's theory of atoms in molecules is often useful for charge analysis.  We have developed computational method for partitioning a charge density grid into Bader volumes which is efficient, robust, and scales linearly with the number of grid points.
We have developed computational method for partitioning a charge density grid into Bader volumes which is efficient, robust, and scales linearly with the number of grid points.