博文
泛基因组(1):突破科赫法则的微生物泛基因组-----缘由、概念、起步
|
从人类遗传学和肿瘤学到植物育种、微生物学和病毒学,许多学科都面临着分析快速增长的基因组数量的挑战。
基因组测序技术的进步使我们能够在比以往任何时候都更大的深度上探索更广泛的生物体。利用丰富的基因组
数据,泛基因组分析已成为研究给定分支基因组中整个基因家族的代表性学科,从而为特定分支的治疗和/或工
程设计提供有效策略。本文提供了对细菌多样性的理解和更新。
1. 突破科赫法则
2. 突破科赫法则的依据
3. 泛基因组概念
4. 细菌基因多样性缘由
5. 日新月异的泛基因组研究
6. 应用
1. 突破科赫法则
1990年前,科赫对急性感染所持的细菌感染的单克隆原则被认为是不可改变的。已建立的单克隆范例可能已经充分描述了急性细菌感染的一个子集,单克隆意味着定植细菌都属于同一基因型【1】。然而,对于大多数细菌感染,尤其是慢性细菌感染,这是一个过于简单的观点。但是,依靠细菌培养的传统诊断方法有助于在医学微生物学中巩固这些范例。
科赫对急性感染所持的单克隆原则在慢性感染方面已被证明是不充分的,因为持久性和慢性细菌感染需要细菌种群中多种形式的异质性。这种对细菌多样性的理解源于当时分子生物学和成像科学中的新技术的综合,如聚合酶链反应(PCR)、先进的代谢分析显微镜技术和全基因组测序(WGS)。这些技术表明,种群或物种内细菌菌株之间存在广泛的表型和基因型多样性【2-4】。感染细菌群体内的基因型多样性表明,这些群体实际上是多克隆的,而不是单克隆的,如,这些种群中同时存在相同细菌种类的多个独立菌株【3-8】]。这种多样性还意味着,即使是单种细菌种群也不是表型克隆或基因型克隆【3】。
2. 突破科赫法则的依据
1995年,科学家发布了第一个基因组图谱:流感嗜血杆菌基因组【9】,1996年发表了真核生物酿酒酵母的全基因组序列【10】,随后大肠杆菌和鼠耳芥等模型生物的基因组也相继破解。截止2005年,公共数据库包含239个完整的细菌基因组。然而,在83%和8%的病例中,每个细菌物种只有一个或两个基因组分别被测序。Tettelin等【11】 的一项研究分析了B组链球菌(GBS)菌株中代表血清群多样性的八个基因组,以回答需要多少基因组才能完整描述一个细菌物种的问题。这项研究发现每个GBS菌株平均含有1806个存在于每个菌株中的基因(core genome,核心基因组]),外加439个在一个或多个菌株中缺失的基因(Dispensable genome,非必需基因组)。非必需基因也分为存在于两个或两个以上但并非所有菌株中的基因(基因组的18%)和每个菌株特有的基因(基因组的1.5%)。基于八个基因组的数学建模表明,即使在数百或数千个基因组测序后,独特的基因仍将继续出现【11,12】。因此,核心基因和非必需基因分别代表了物种的本质和多样性。
就GBS而言,泛基因组(Pan-genome)包含2713个基因,其中1806个属于核心基因组,907个属于非必需基因组。每次对新菌株测序时,预计GBS pan基因组平均增长33个新基因(图1)。对五株化脓性链球菌进行的类似分析【11】显示了相似的基因组多样性,表明每增加一个新基因组,就有27个特定基因的渐近值,再次导致“开放”泛基因组(‘open’ pan-genome.)。在八个独立炭疽杆菌分离物的研究中观察到不同的行为。在这种情况下,在只添加第四个基因组后,发现添加到泛基因组的特定基因数量迅速收敛到零【11】。因此,炭疽杆菌物种有一个“封闭”的泛基因组(‘closed’ pan genome),即四个基因组序列足以完全描述该物种(图1)。也就是说,Tettelin等【11】提出了预测泛基因组大小的方法:如果每检测一个新菌群后出现新的基因数目趋于稳定,说明泛基因组是有限的。如果每检测一个新菌群后出现的新的基因数目千差万别,说明泛基因组是无穷大的(图1)。
在对大量菌株进行测序后,开放泛基因组中非必需基因数量比核心基因组的大小大几个数量级,迫使我们重新考虑细菌物种的定义【11,12】。


图1。与一个物种或物种泛基因组相关的一组基因取决于可用基因组序列的数量。在该图中,无乳链球菌(红点)
和炭疽杆菌(蓝色三角形)泛基因组的大小显示为测序菌株数量的函数。一个物种泛基因组的大小可以随着测序
菌株的数量而增长,或者很快饱和到一个极限值。无乳链球菌泛基因组是“开放的”;炭疽杆菌已“关闭”。
许多间接证据已经暗示了泛基因组的概念,甚至在它被数学量化正确定义之前【11】。几项使用同一物种的多个分离物进行的消减杂交和比较基因组杂交(comparative genome hybridization,CGH)研究表明,幽门螺杆菌、金黄色葡萄球菌和大肠杆菌等细菌物种显示出广泛的遗传多样性,平均20–35%的基因对单一菌株具有特异性【12-15】.这些物种中存在如此多的菌株特异性基因表明——就像GBS的情况一样——它们也可能显示一个开放的泛基因组。这就提出了一个问题,即微生物世界是否包含足够的基因来预测泛基因组产生的如此巨大的基因库。
3. 泛基因组概念
第一个基因组图谱--流感嗜血杆菌基因组发表十年后的2005年,基因组学如何描述细菌物种的问题尚未完全解决。实验数据表明,在某些物种中,甚至在对几个菌株的基因组进行测序后,也发现了新的基因。数学模型预测,即使对每个物种的数百个基因组进行测序,也会发现新的基因。这些结果表明,不同群体之间的基因组存在不可忽略的差异。于是,提出了泛基因组这个概念,通过检测群体之中每个个体的基因组,综合起来就称为泛基因组。因此,细菌物种可以通过其泛基因组来描述【11,12】。
2000年,Sigaux首次使用术语“泛基因组(Pan-genome)”来描述一个公共数据库,该数据库包含对主要类型肿瘤、组织和实验模型的基因组和转录组改变的评估【16】。
2005 年,美国马里兰大学的 Tetterlin 教授等在研究无乳链球菌的多种不同致病菌株时提出泛基因组(Pan-genome)概念 (pangenome,pan源自希腊语‘παν’,全部的意思)【11】。他当时描述为,共享于所有菌株的 “核心基因组(core genome)” 和分散在单个或者部分菌株基因组中的差异序列,即,“非必需基因组(Dispensable genome)” ,根据这一定义,泛基因组捕获了一个物种的全部基因内容。最初,在泛基因组中区分了两个部分,一个是核心基因组,代表所有菌株之间共享的一组基因,另一个是非必需基因组,由菌株特异性基因或菌株子集之间共享的基因组成。然而,通常使用的泛基因组划分(参见图 2) 区分三个主要部分:核心基因组、非必需基因组或辅助基因组(accessory genes)和单体基因组(singleton genome)【17】。

图2。 三个基因组A、B和C的泛基因组。
核心基因组以黑色显示,白色交叉点代表非必需基因组,灰色部分代表单体基因组
非必需基因组(dispensable / variable / accessory genome)(图3)通常进一步细分为一个菌株特有的基因(称为“独特基因,unique genes”)和一些但并非所有菌株之间共享的基因(称为“辅助基因,accessory genes”)(图4a)【18】。用基因而不是DNA序列来定义泛基因组对原核生物来说是明智的。在这些物种中,基因不仅包含了大部分(通常为90%或更多)的序列内容,而且基因内容变化很大;在一些细菌物种中,已发现独特的基因占泛基因组的20%至40%。存在哪些基因的这些差异通常有助于致病性、耐药性和人类健康感兴趣的其他表型;因此,分析非必需基因组与核心基因组可以帮助解释这些表型。

图3。核心基因组,代表所有菌株之间共享的一组基因;
非必需基因组(dispensable / variable / accessory genome)仅在某些个体中存在。

图4。核心基因组和非必需基因组
1a细菌和其他原核生物基因组主要由基因组成,很少有基因间序列。一个物种的核心基因组由所有菌株共享的基因组成。
非必需基因组由一些但并非所有菌株(辅助基因)共享的基因和仅存在于一个菌株(独特基因)中的基因组成。
核心基因组和非必需基因组共同构成泛基因组。
2b真核生物基因组的基因含量变化不大。泛基因组包括基因间的序列以及基因。
核心基因组为一个物种的系统发育提供了基因组学基础,并被认为在不同的分类水平上具有代表性【19】。非必需基因组可能包含补充的生化途径,提供一些选择性优势,如环境适应、特定毒力或抗生素耐药性【20,21】。由于不同菌株的基因组是可用的,研究可能针对与物种进化相关的问题,通过比较分析阐明物种的基因组多样性。存在于致病物种所有菌株中的基本基因可以用作潜在的抗生素靶点【22】。
核心基因组由负责物种代谢基本方面和主要表型特征的“基本”基因组成【23,24】,包括维持功能的基因,如能量生产、氨基酸代谢、核苷酸代谢、脂质运输和翻译机制【9,25】。相比之下,非核心(分布的、辅助的或适应性的)基因组包括编码补充或修饰的生化功能的基因,这些功能可能在基本生存以外的环境中有用,例如适应新环境、抗生素耐药性或新环境和宿主的定殖【23,24】。非核心意味着这些基因可以从基因组中删除,但这种删除可能导致重要表型特征的丧失,如在非传统营养物质和基质上生长的能力、毒性和抗生素抗性【23,24】。早期研究表明,一个物种超基因组中的大多数非核心基因与该物种的核心基因一起进化【26】,并且许多未注分布的基因与在不同环境中的生存相关【9】。非核心基因也包括那些具有寄生功能的基因,即自私基因。这些基因包括促进自身转移和繁殖的基因,以及运行“保护网”(毒素-抗毒素基因)的基因【27】。 在对2005年至2019年发表的295个物种特异性超基因组项目的分析中,发现基本上在所有情况下,超基因组比核心基因组大得多,核心基因组占超基因组的比例从<20%到>60%【25】,随着鉴定新基因的个体数目的增加,泛基因组的大小也随之增加,但核心基因的百分比随之下降(图5)。有趣的是,基因组和超基因组的大小都与物种生物学有关。自由生活的环境细菌物种往往具有最大的基因组4-12百万碱基(Mb);具有中等大小基因组1.5-4 Mb的共生菌和致病菌,专性和细胞内病原体的基因组最小为0.6-1.5 Mb,然而,这些规则并不严格,因为也有例外,因为并非所有病原体的基因组都缩小了【25】。
结构变异中的存在/缺失变化(PAVs: Present/absent variations)是泛基因组的重点研究对象。

图5. 显示不同细菌种类的核心大小和超基因组之间关系的示意图。
每个物种的内圈面积代表核心基因组相对于超基因组的大小,超基因组由圆圈的整个面积表示。所有区域都代表核心
基因组和超基因组之间的实际百分比差异。从左下角结核分枝杆菌开始,顺时针穿过金黄色葡萄球菌、珊瑚球菌、
大肠杆菌和铜绿假单胞菌,与超基因组相比,核心基因组的相对大小减小。
4. 细菌基因多样性缘由
16S RNA测序和脉冲场凝胶电泳(PFGE)分析,在同一物种中发现了多达1287种不同的基因型,其中大多数通过插入或删除大型基因组元素来区分。虽然新基因可以通过复制现有序列,然后多样化而产生,但获得新功能的最常见方式是从无关生物中转移遗传物质。这项研究提供了有关细菌种群遗传多样性的有启发性的数据,清楚地表明,在单一生态系统的看似同质的种群中,遗传内容的宏观差异也存在【28】。
这些技术表明,细菌具有复杂的生命周期、多微生物生态,并通过基因特征的水平交换进行原位进化。因此,在感染过程中不断产生多样性,导致微生物群落比以前想象的要复杂得多(图6)。这一观点基于一个基本原则,即感染群体中的细菌表现出基因多样性,包括基因拥有差异,这是由水平基因转移机制(包括转化、接合和转导)造成的。这种理解体现在超基因组/泛基因组和分布式基因组假说(DGH)的概念中。这些范式促进了细菌生态学不同领域的多种研究,包括宿主-细菌相互作用,涵盖了共生关系的范围,包括互利共生、共栖和寄生【9】。
自从1977年Sanger和Coulson引入DNA测序以来,已经取得了很大的进展。产生的数据量越来越大,需要不断先进的计算机处理。在不同的领域,已经发表了许多阐述不同方法的研究,例如基因组组装和注释,以及对新细菌物种和细菌分类的研究【29】。2004年,Venter等首次将“全基因组鸟枪测序技术”应用于环境样本。它大大提高了我们对未培养细菌世界的复杂性和易变性的理解【30】。一项研究表明,肠道菌群中的物种数量远高于预期,不同地点和不同受试者之间存在相关差异【31】 。这就提出了这种遗传多样性的起源问题。细菌可以通过接合、转化和噬菌体感染(转导)从环境中获取基因【32】,对环境样本的实验研究表明,单个生态系统中可用的遗传物质数量足以构成几乎无限的新基因库。迄今2005年,在所有这些多样性中,只有不到1000个微生物基因组被完全测序(图6)。
这里描述的惊人的大基因库表明,在进化过程中,绝大多数新功能可能是在微生物世界中产生的,而不是在大型动物(如人类)中产生的,因为人类只有25000到35000个基因。这将导致微生物和大型动物在进化中扮演完全不同的角色。事实上,在这一理论下,微生物会产生新的基因和功能模块,而大型动物则会首先利用微生物产生的模块,然后在基因组内以多种不同的方式重新排列它们,并通过mRNAs的选择性剪接进行进化【23】。

图6。微生物世界的多样性。
原核物种的遗传多样性包含两种不同的现象:(1)遗传异质性,即群体中的个体拥有不同的等位基因(同一基因的变异形式);(2)基因组可塑性,即群体中的个体拥有不同的基因【33】。因此,基因组可塑性准则告诉我们,一个物种中一个菌株的基因组不能解释整个物种的整套基因,对单个分离物的研究将大大低估物种的生物学特性。因此,需要对多个独立分离株的基因组进行充分表征,以估计细菌物种的总生物复杂性【11,22,23,34,35】。总体而言,一个物种内所有个体的基因组中包含的基因构成该物种的超基因组(或泛基因组)【11】。
细菌基因组可塑性的特点是,群体中的每个菌株都拥有群体超基因组中一组独特的分布/辅助基因【22,23,33】。现在已经证明了多个物种的高度基因组可塑性,这是观察到这些物种同时具有致病性和共生菌株的基础,这些菌株包括:流感嗜血杆菌【36】、铜绿假单胞菌【37】、肺炎链球菌【34,36,38】、卡他莫拉菌【39】等。基因组多样性源于基因组扩展(增加基因)和基因组减少(删除基因)之间的平衡【22】。将流感嗜血杆菌(Rd)和肺炎链球菌(Sp6)的类型菌株与临床分离株(各自物种)进行比较,结果显示,基因组大小变化较小,但每个基因组对的比较(平均)超过200个指标,表明发生了非常广泛的重组【38】。
基因组可塑性是通过三种水平基因转移(HGT)过程产生的【1,12】:(i)通过转化(transformation),当遗传物质可以从环境中获取时;(ii)当DNA由病毒传递时,通过转导(transduction);(iii)当DNA在细胞间直接交换时,通过接合或共轭(conjugation)(图7)。转化和接合需要源生物体和目标生物体紧密接触,而噬菌体可能使居住在不同环境中的细菌物种能够交换遗传物质,而遗传物质通常包含对发病机制至关重要的基因。微生物世界中存在的全部基因库可能超过迄今为止所作的任何估计数几个数量级,数十亿基因的存在不再是意料之中的(图6)。
垂直基因转移是一种标准的机制,通过这种机制,母细胞复制其整个DNA补体,并在细胞分裂期间将每个染色体和质粒/上位体(上位体通常被描述为能够整合到主要细菌染色体中的质粒)的相同(或几乎相同)拷贝传递给每个子细胞 。相比之下,HGT过程涉及两个通常不相关的细菌细胞之间的单向基因运动,其中一个或多个供体染色体DNA片段(范围:几百个碱基至>100个碱基) Kb)(和/或接合情况下的质粒和外体DNA)转移到受体细胞,导致受体细菌染色体的部分替换或获得新的复制子【33】(图7)。

图7。演示不同类型细菌遗传的示意图。
(A) 细菌细胞,具有蓝色的单环状染色体;
(B) 从母细胞到子细胞的垂直传播,子细胞彼此和母细胞在基因组上相同;
(C) 用红色阴影表示的转移或可转移DNA的水平基因转移方法:
(i)转化,有能力的细菌从环境中提取外源DNA,然后通过同源重组将其重组到细菌染色体中;
(ii)接合,通过菌毛将质粒(上位体)或染色体DNA从“雄性”寄生细菌转移到受体;
(iii)转导,利用非同源重组通过噬菌体转移外源细菌DNA。
重要的是要理解,转移DNA来源的细胞在转移时不一定必须是活的(在转导的情况下也不一定是活的)。每种HGT机制都可能发生在同一物种的不同菌株之间以及相关物种之间。然而,接合,特别是当它涉及到上位体转移时,也可能发生在存在于不同门中的非常不同的物种之间【40】。随着遗传异质性水平的增加,转化率迅速下降,因为它们依赖于受体细胞内的同源重组机制【41,42】。转导基于感染温和噬菌体的宿主范围,因此通常仅限于单个物种或密切相关的姊妹物种。
一些细菌使用单一HGT过程,而另一些细菌使用两种甚至全部三种(图7)。例如,金黄色葡萄球菌主要依赖于转导,因此其泛基因组比许多其他细菌物种小得多【36】。相反,大肠杆菌利用交配和转导,而流感嗜血杆菌利用所有三个HGT过程【42】。交配和转化是一个活跃的过程,要求供体和受体生物体彼此生活在非常接近的地方。这两个过程都需要一个或另一个参与细菌消耗大量能量。因此,供体或受体细菌,以及间接参与这些过程的细菌种群,必须从这些过程中获得进化优势,否则它们不会持续存在。根据定义,任何需要能量消耗的机械过程都必须为其持续传播提供进化优势。在交配的情况下,主要的能量消耗是通过雄性(DNA供体)细菌进行的。这一过程被描述为寄生过程,是自私基因确保自身繁殖的一个例子。这是因为交配器的基因是转座因子的一部分(位于染色体或上位体)这些基因共同确保了它们自身的水平繁殖,同时也转移了物理上相邻的基因,这些基因因其在压力环境中为宿主细菌提供生存优势的能力而被选中。因此,交配通常也会导致基因转移,从而提高受体的存活率,这表明一个单一的过程可以产生多种生态结果。在交配过程中,两个活细菌通过菌毛(或类似结构)暂时连接在一起,其中一个细菌通过菌毛将其DNA(染色体和/或外染色体)的拷贝发送到另一个细菌中。因此,在供体(自私基因)自我繁殖的过程中,进化优势会累积到供体(自私基因)身上,而在受体获得能够生存的基因(如编码抗生素和重金属抗性的基因)的过程中,进化优势也会累积到供体(自私基因)身上。出于这些原因,我们将这些HGT过程称为群体级毒力(或存活)特征。
细菌HGT机制通过提供“进化捷径”,使生物体能够快速适应变化或新环境【23】,并通过提供突变等位基因的替换[34],有助于确保种群在环境和营养挑战期间生存。HGT还可以诱导/产生受体细胞的主要生活方式改变,并允许辐射进入不同的生态位【22】,综上所述,这些观察结果表明,进化压力选择了产生多样性的机制【1】。
5. 日新月异的泛基因组研究
随着1975-77年第一代测序技术的出现【43,44】,以及2004年高通量测序技术的出现【30,45】,获得完整基因信息的途径发生了深刻的变革。得益于这些现代高通量测序技术,产生了大量数据,使基于泛基因组分析的研究成为可能(图8)。在高通量测序时代刚刚开始之后,Tettelin等人【11】于2005年提出了泛基因组的第一个定义。泛基因组可以定义为属于研究组的整个基因内容【4,46,47】。其应用是多方面的,包括致病性研究【48,49】,移动组【50】,抗性组【51】,细菌生活方式预测【29】,以及分类学研究。事实上,泛基因组研究允许对物种进行重新分类【52】,从而澄清和改进先前提出的传统标准【29】。

图8。PubMed网站基因组数据库中所有泛基因组研究每年的出版物数量
(https://www.ncbi.nlm.nih.gov/pubmed)
测序技术的最新进展减少了对整个细菌基因组进行测序的时间跨度和成本。高通量下一代测序(NGS)技术产生了大量关于微生物种群的数据,这些数据可在不同的存储库中公开获取(图9)。因此,从进化、生态学和多样性的角度研究和比较一个物种或属内不同细菌菌株的基因组成为可能。对泛基因组的研究为破译一个物种的毒力和发病机制的微进化、全部组成和多样性提供了见解【53,54】。
基因组学的大部分领域都围绕着参考基因组的存在,参考基因组是每个物种“典型”个体的路线图。每个参考基因的创建过去是,现在仍然是基因组学界的一个主要关注点. 第一批泛基因组是为小型、易于测序的细菌开发的,但即使在这种情况下,泛基因组也提供了新的科学见解【11】。考虑细菌物种内的遗传多样性有助于我们理解致病性、毒力和耐药性的潜在差异,甚至有助于预测新菌株的致病性. 仅人类参考基因的创建就花费了13年和27亿美元。将新测序的个体与参考个体进行比较并发现差异的能力促成了无数的发现和创新,在人类基因组学中,这一能力已成为数千项寻找疾病遗传起源的研究的基础【55】。
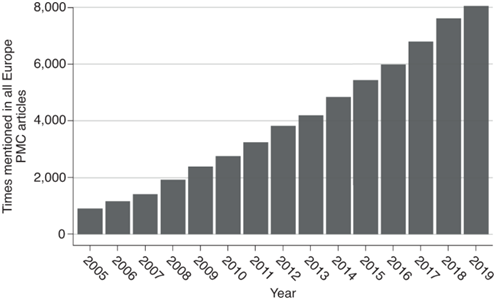
图9。Number of times the terms ‘pangenome’ or ‘pan-genome’ are mentioned in Europe Pubmed Central from the first mention in Tettelin et al.1.
“泛基因组”或“泛基因组”一词自Tettelin等人首次提及以来,在欧洲公共医学中心被提及的次数。
6. 应用
泛基因组测序是运用高通量测序及生物信息分析手段,针对不同亚种/个体材料进行测序,分析泛基因组特征,不仅可以获得多个基因组,完善该物种的基因集,还可以获得种群甚至个体特有的DNA序列和功能基因信息,为系统进化分析及功能生物学研究奠定基础。
泛基因组分析能鉴定出参考序列中不存在的基因,对这些新基因进行功能分析也是其研究的焦点。
泛基因组分析得到了大量核心基因和可变基因,例如,可以挖掘发现每一个个体中独特的基因。这些基因都行使着什么样的功能,以及相互之间有什么样的联系?这些问题将在未来泛基因组的研究中得到解答。可变基因的作用之一是提供适应环境变化和新的生态位所需要的表型可塑性,为了履行这一功能,可变基因需要整合到现有的生物学通路和调控网络中。
泛基因组可以应用到全面准确的变异检测: 与重测序变异检测相比,泛基因组研究基于基因组序列进行变异分析能够大幅度提高变异检测的准确性,以及大结构变异的检出性(SNP、Indel、CNV、PAV)。在群体中各个个体相对差异较大时,使用PAV分析比使用SNP等变异信息来分析更加能够体现群体内部的差异, 也能看作为其中一种marker,基于PAVs也能对物种进行遗传进化的关联分析。泛基因组对PAV的分析大多集中在编码区域,然而,越来越多的人认识到顺式调控元件和重复序列在健康和疾病中的重要性。因此,泛基因组的研究也扩展到非编码序列。
主要参考文献
1. Biochem Soc Trans. 2020 Dec 18;48(6):2437-2455.
2. J. Appl. Biomater. Funct. Biomater. 2014,12, 13–20
3. Clin. Orthop. Relat. Res. 2005,437, 20–24
4. Fut. Med.2008, 3, 31–42
5. Epidemiol. Infect. 1996,116, 177–183
6. Infect. Immun.1997, 65, 1468–1474
7. J. Infect. Dis. 1999,180, 404–409
8. FEMS Immunol. Med. Microbiol. 2010,59, 269–279
9. Science 1995;269(5223):496–512
10. Science 1996;274(5287):546–67.
11. Natl Acad Sci USA 2005;102(39):13950–5.
12. Curr Opin Genet Dev . 2005 Dec;15(6):589-94.
13. Proc Natl Acad Sci USA, 98 (2001), pp. 8821-8826
14. Genome Res, 11 (2001), pp. 1706-1715
15. J Bacteriol, 186 (2004), pp. 3911-3921
16. Bull Acad Natl Med 2000;184(7):1441–9.
17. Methods Mol Biol. 2018;1704:29-53.
18. Nat Rev Genet. 2020 Apr;21(4):243-254
19. Proc Natl Acad Sci USA 2005, 102(Suppl 1):6595–6599
20. Curr Opin Microbiol 2006, 9(5):496–498
21. Curr Opin Microbiol 2015, 23:148–154
22. Int Microbiol 2010,13(2):45–57
23. Curr. Opin. Gen. Dev. 2005,15, 589–594
24. Int. Microbiol. 2010,13, 45–57
25. Innamorati, K.A., Earl, J.P., Aggarwal, S.D., Ehrlich, G.D. and Hiller, N.L. (2020) The bacterial guide to designing a diversified portfolio In The Pangenome (Tettelin H. and Medinie D., eds.), pp. 51–88, Springer, Cham, Switzerland
26. Nucleic Acids Res. 33, 3644–3658
27. PLoS Genet. 2009,5, e1000437
28. 20. Science, 307 (2005), pp. 1311-1313
29. Biol Direct . 2019 Feb 26;14(1):5.
30. Science 2004, 304:66-74.
31. Science 2005, 308:1635-1638.
32. Microbiol Mol Biol Rev 2004, 68:560-602.
33. Clin. Orthop. Relat. Res.2005, 437, 20–24
34. Infect. Immun. 2006,74, 321–330
35. J. Bacteriol.2012, 194, 3922–3937
36. BMC Genom.2011, 12, 187
37. Environ. Microbiol. 2012,14, 2200–2211
38. J. Bact. 2007,189, 8186–8195
39. Genome Biol. Evol. 2016,8, 955–974
40. Mol. Biol. Evol. 1998b,15, 385–390
41. Science 2007,315, 476–480
42. J. Bacteriol. 2014,196, 1471–1483
43. J Mol Biol. 1975;94:441–448.
44. Proc Natl Acad Sci. 1977;74:5463–5467.
45. Indian J Microbiol. 2016;56:394–404.
46. Curr Opin Microbiol. 2008;11:472–477.
47. Int Microbiol Off J Span Soc Microbiol. 2010;13:45–57.
48. N Engl J Med. 2011;365:709–717.
49. Proc Natl Acad Sci U S A. 2009;106:15442–15447.
50. BMC Genomics. 2010;11:688.
51. Front Microbiol. 2013;4:103.
52. Biol Direct. 2015;10:55.
53. Adv Exp Med Biol . 2018;1052:39-49.
54. Nat Plants. 2020 Aug;6(8):914-920.
55. Can J Microbiol. 2021 Aug;67(8):553-571.
https://blog.sciencenet.cn/blog-526326-1310902.html
上一篇:传承与创新(2)孔子节的纪念:孔道与孟道/变与不变
下一篇:培养组学(2):人类微生物群培养的取样和保存