博文
痛并快乐着:蛋白质结构预测的边角故事
 精选
精选
||
导读:2020年底,AlphaFold2在蛋白质结构预测的CASP比赛中实现了高精度预测,使得一个平时默默无闻的小众学科变成了家喻户晓的明星。AlphaFold的成功是结构预测这个领域多年来的量变所导致的质变。其中二个重要的量变是从预测分类转变到预测连续的边和角(原子间距离和二面角)。这里,我主要讲讲首先实现的连续二面角的预测,以及它是如何触发了端到端的蛋白质结构预测这个连锁反应,并最终导致了革命性突破的这个故事。
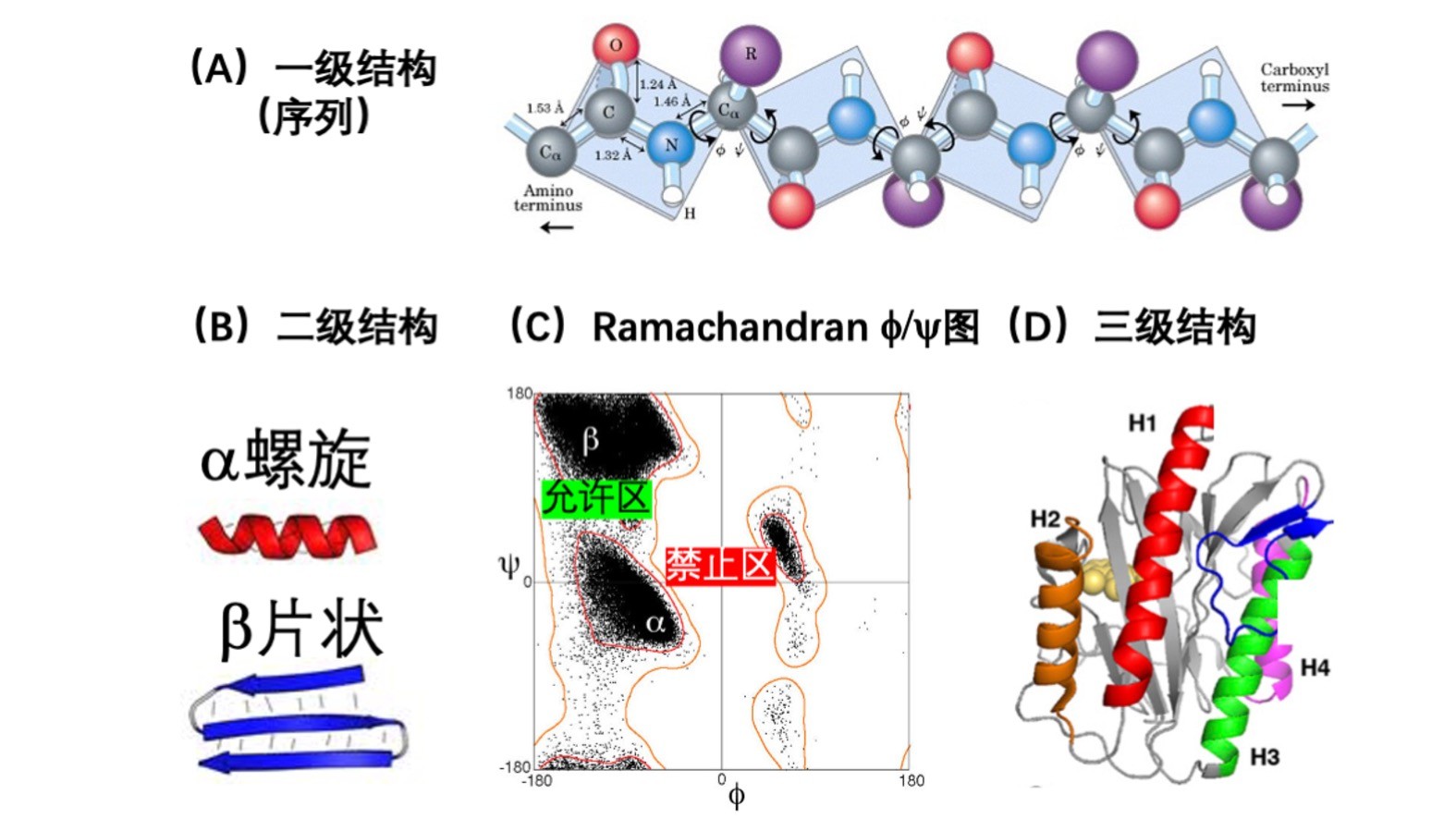
图一:蛋白质的一级结构(A)、二级结构(B)、二面角的角度分布(C)和三级结构(D)的示意图。
蛋白质的氨基酸序列被称为蛋白质的一级结构(图一A),而序列所构成的肽主链的结构构象通常被称为二级结构。从结构形状来看(图一B),它大体可以分为三类:有规律的alpha-螺旋状(helix)和beta-片条状(strand/sheet),以及无规线圈状(coil)。还有一种分法是根据主链的二面角来分。蛋白质主链的结构完全可以通过三个二面角phi, psi,和omega来重建(图一A)。而omega在绝大多数情况下是接近180度(平面)的,所以一般只需要phi和psi就足以决定主链的精细结构了,但是由于支链和主链原子之间的排斥,一些phi和psi角度是禁区,导致分布极不规则(著名的Ramachandran图,图一C)。历史上有一些研究把对应二级结构形状的角度进行分区,但是预测这些无明显规律的角度区域,与预测二级结构形状相比,没有明显的优势,所以自从1958年以来,二级结构的预测一直以预测三态:alpha-螺旋,beta-片条状,和无规线圈为主,这样粗略化的二级结构是无法用来重建蛋白质主链结构的。
我于2000年来到纽约州布法罗大学做助理教授,不久我们组的博士后周宏毅就发展了一个基于模板的蛋白质结构预测方法SPARKS和SP3,并在2004年侥幸获得了国际蛋白质结构预测比赛(CASP)中基于模板预测的第一名(Zhou & Zhou, 2004,2005A,2005B)。要进一步改进SPARKS方法就需要一个更加准确的二级结构预测来帮助搜索更匹配的远源结构模板,于是,我把这个任务交给了我们组新来的、有计算科学背景的以色列博士后Ofer Dor。他通过优化普通神经网络使预测的二级结构达到了80%的准确度(当时最高)(SPINE, Dor & Zhou, 2007A)。在做这个项目过程中想到,为什么不绕过粗略的三态二级结构,而直接去预测连续的二面角呢?也就是说把分类的问题(classification)变成回归的问题(regression)。于是就先试了一下psi角,发现由于角度的周期性没有处理好(-180°=+180°),预测误差太大,平均54°(Real-SPINE, Dor & Zhou, 2007B),我才明白过来:原来是自己初生牛犊不怕虎,想法太天真,难怪大家都避开直接预测连续的角度。
2006年,我离开纽约州的布法罗来到了印第安纳波利斯市的印第安纳大学做正教授,Ofer也回以色列创业去了。虽然之前对psi角度预测不算成功,我仍旧不死心,就让生物物理专业出身的薛斌博士后继续这个课题,并将psi和phi一起预测,可以用来直接构建主链结构。他发现,通过对psi角度的简单位移,让不容易预测的两端(-180°,+180°)移在角度分布少的地方,就可以把角度的误差一下子从54°降到38°,而phi的误差为25°(Real-SPINE2, Xue et al, 2008)。据我们所知,这是世界上第一个同时预测psi和phi真实连续角度的方法,并有相当的准确度。
不久Eshel Faraggi博士和张社生博士加入团队,他们通过多态预测与真实角度预测的结合以及神经网络算法的改进,成功地把psi角的误差进一步下降到33°。同时杨跃东博士利用他们所预测的连续角度和三态二级结构,证明了在预测三级结构中,连续角度是比粗略化的三态二级结构要好得多的约束,因为预测的角度有无规线圈区内的有用信息(图二,SPINE XI,Faraggi et al, 2009)。在当时,几乎所有比较成功的蛋白质结构从头预测方法都是通过结构碎片或者模板的组装来预测三级结构的(例如David Baker组的Rosetta,Skolnick,张阳组的TASSER,I-TASSER,许锦波的Raptor X,我们组的SPARKS X(Yang et al, 2011)等等),而我们通过预测真实角度来建立、约束、和能量优化主链结构,完全不需要用蛋白质的已知结构或者已知结构碎片来作为模块,从而开创了一条新路(Zhou et al, 2011)。
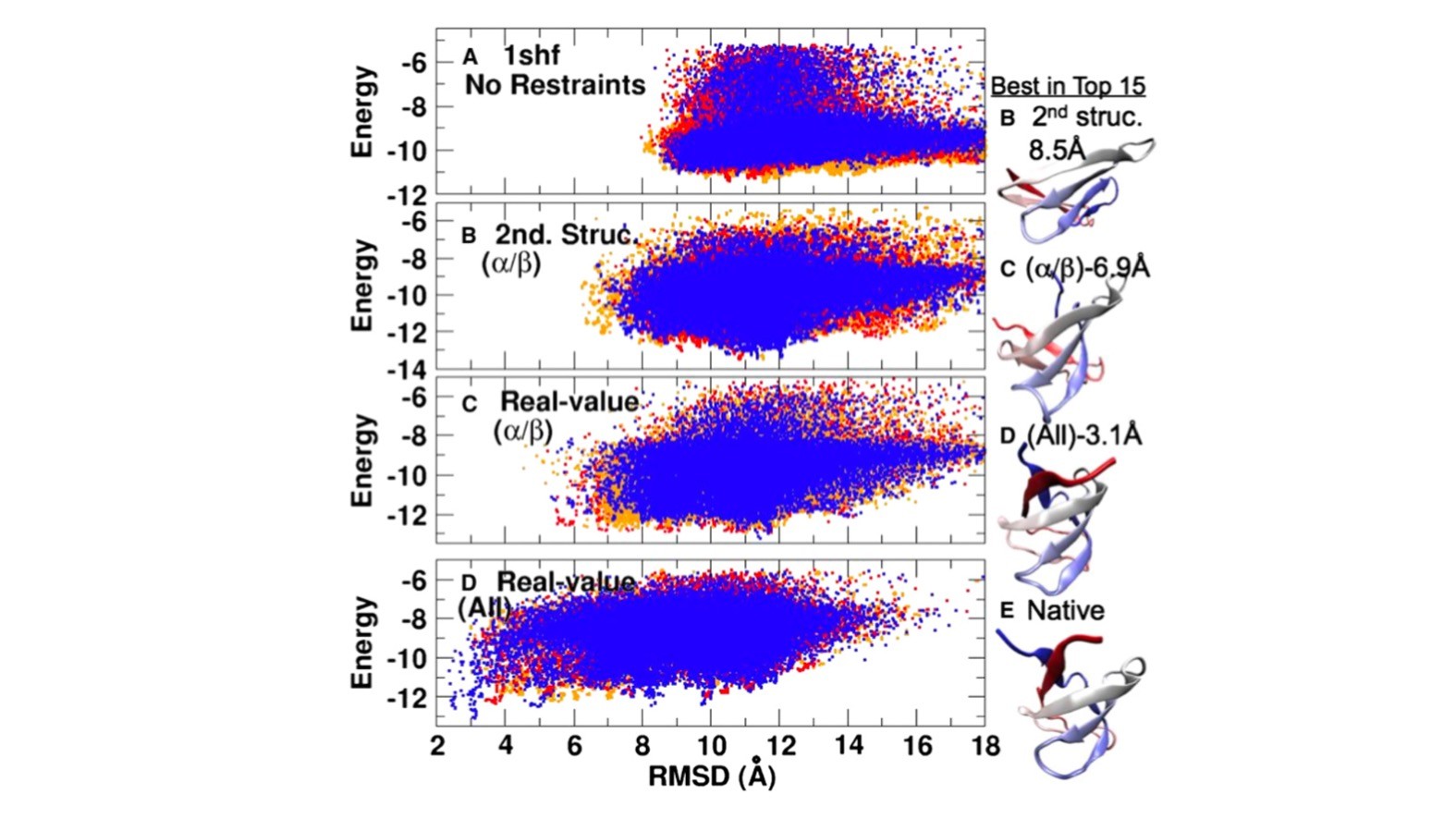
图二:预测的三级结构能量与结构准确度(RMSD)的关系:在不用约速(A)、用预测的有规律二级结构作为约速(B),用预测的、有规律二级结构区的角度作为约速(C),和用所有预测角度作为约速(D)所得的结果。这个结果表明:在无规线圈区所预测的角度对获得高精度结构(小RMSD)及其重要(摘自Faraggi et al, 2009),该结果在发表在2009年Structure后并没有能很快改变大多数人只利用预测的二级结构来约速三级结构的习惯。
2013年,我来到澳大利亚格里菲斯大学,我们组开始和格里菲斯大学的Kuldip Paliwal教授合作,使用不同深度的深度学习方法来进一步改进角度的预测。Paliwal是国际著名的机器学习专家,他和他的同事(Dr. Schuster)是第一个提出被广泛应用的双向循环神经网络(Bidirectional recurrent neural network)。我们的第一篇合作论文(Lyons et al, 2014)第一次将深度学习应用于蛋白质连续角度预测,并把角度的直接预测改成先预测SIN和COS,再通过计算ARCTAN来得到真实角度,从而避开了角度的周期性。psi角的误差从33°降到30°(3层隐藏层的SPIDER2,Heffernan et al, 2015),到27°(4层隐藏层的长短期记忆双向循环神经网络SPIDER3,Heffernan et al, 2017),到23°(>10层隐藏层的SPOT-1D,Hanson et al, 2019),而phi的误差也最终降到了16°;与此同时,二级结构的预测也接近了理论的极限(准确度86%)(SPOT-1D,Hanson et al, 2019)。也就是说,通过这十几年的努力(2007-2019),我们把真实角度的预测从几乎无用的精确度变成了可以直接用来构建越来越可靠的主链结构(SPOT-1D,Hanson et al, 2019,图三)。

图三:直接用预测的角度构建的不同类型的、40-氨基酸长的主链结构(摘自Hanson et al, 2019)。
用预测的连续角度来直接构建主链结构、用能量函数来优化和预测三级结构:这是我们提出的、无结构碎片结构预测的设想(Fragment-free structure prediction, Zhou et al, 2011)。但是这个设想并没有能走太远,因为即使有好的二面角,没有高精度的能量函数去导向准确的结构,还是不行。事实上,蛋白质结构从头预测的所有方法一直进展很慢,都是被没有好的能量函数所拖累的。缓慢的进步靠的是越来越准确的二级结构和距离接触图的预测,并用这些预测来约速、改进不正确的能量函数的导向而实现的。2019年,AlQuaris把我们的无结构碎片结构预测这个设想(二面角预测-->主链结构构建-->能量函数优化)全部搬到深度学习的神经网络内(二面角预测-->主链结构构建-->结构误差反馈,AlQuarishi, 2019)。这个端到端的、从主链二面角到蛋白质三级结构的训练和预测,是一个重要的转折点,它第一次表明,能量函数的作用完全可以在神经网络内部实现,而神经网络里几乎无限量的参数,与几十或者几百人工经验参数的经典力场或者经验能量函数相比,有更好的能力来模拟复杂的蛋白质内部的相互作用。由于没有利用支链紧密堆积的信息,也没有利用共进化的距离信息,这个方法在后来实际蛋白质结构预测CASP比赛表现中并不突出。
尽管如此,AlphaFold2明显是受到了AlQuarishi进行端到端结构预测的启发。不过它在这个基础上有新的创新:不再仅仅考虑粗粒化的主链,而是首先预测氨基酸残基的位置和氨基酸支链的二面角,以及残基之间的直接距离,再通过优化把残基连接起来、构成主链。这个创新抓住了蛋白质结构的稳定性是靠疏水支链的紧密堆积这个主要矛盾,避免了局部最优,实现了蛋白质结构预测的革命性突破(Jumper et al, 2021)。我们注意到AlQuaris和AlphaFold2中主链或者支链二面角的预测也是通过SIN/COS到ARCTAN的变换来避免角度的周期性的。值得一提的是,从离散的二态接触图预测到连续的接触距离预测,许锦波教授在这方面作出了贡献。
综上所述,AlphaFold2在蛋白质结构预测上革命性的成功,是在点点滴滴的积累以及一环扣一环的进步之后才能一跃而成的。其中,从分类到连续真实二面角的预测是重要的一环,共进化信息所导致的精确连续距离的预测则是平行的另外一环,而由连续真实二面角构建蛋白结构所启发的、摆脱了能量函数的端到端预测则是关键点。综合这些阶段性成果,加上先支链后主链的预测是最后的临门一脚。
蛋白质真实、连续的二面角预测,作为一个研究方向,一直是冷冷清清的、没有引起太多同行的关注,我完全凭个人的兴趣在坚持着,即使到现在也只有少数几个他人发展的真实二面角预测方法,与有超过300个预测蛋白质二级结构的方法比(Yang et al, 2018),可以说是天上地下。虽然在当时、甚至在现在,并不能一下子让人看出预测连续真实二面角的重要性。可以说,不被理解和接受是做原创的痛点,但一旦直接或者间接成就了未来的突破,它是快乐的源泉。痛并快乐着,这就是科研人的真实写照。
感谢密苏里大学许东教授和中山大学杨跃东教授的阅读和建议。也特别感谢中国人民大学龚新奇教授的讲座邀请,促使了这篇文章最后的结稿。
AlQuraish,M. (2019). End-to-End Differentiable Learning of Protein Structure, Cell Systems, 8, 292-301.
Dor,O. and Y. Zhou, (2007A), Achieving 80% ten-fold cross-validated accuracy for secondary structure prediction by large-scale training, Proteins 66, 838-845.
Dor,O. and Y. Zhou, (2007B). Real-SPINE: An integrated system of neural networks for real-value prediction of protein structural properties, Proteins 68, 76-81.
Faraggi, E.,Y. Yang, S. Zhang and Y. Zhou, (2009). Predicting continuous local structure and the effect of its substitution for secondary structure in fragment-free protein structure prediction, Structure 17,1515-1527
Hanson, J.,K. Paliwal, T. Litfin, Y. Yang, and Y. Zhou (2019). Improving prediction of protein secondary structure, backbone angles, solvent accessibility, and contact numbers by using predicted contact maps and an ensemble of recurrent and residual convolutional neural networks, Bioinformatics, 35: 2403–2410.
Heffernan, R.,K. Paliwal, J. Lyons, A. Dehzangi, A. Sharma, J. Wang, A. Sattar, Y. Yang and Y. Zhou, (2015). Improving prediction of secondary structure, local backbone angles, and solvent accessible surface area of proteins by iterative deep learning, Scientific Reports, 5 11476.
Heffernan, R.,Y. Yang, K. Paliwal, and Y. Zhou, (2017). Capturing non-local interactions by long short term memory bidirectional recurrent neural networks for improving prediction of protein secondary structure, backbone angles, contact numbers, and solvent accessibility, Bioinformatics, 33: 2842-2849.
Jumper,J. et al. (2021). Highly accurate protein structure prediction with AlphaFold, Nature, 596, 583-589.
Lyons, J. A. Dehzangi, R. Heffernan, A. Sharma, K. Paliwal, A. Sattar, Y. Zhou, and Y. Yang (2014), Predicting backbone Cα angles and dihedrals from protein sequences by stacked sparse auto-encoder deep neural network, J. Comp. Chem. 35, 2040-2046.
Xue, B.,O. Dor, E. Faraggi and Y. Zhou, (2008). Real value prediction of backbone torsion angles, Proteins 72, 427-433.
Yang, Y., E. Faraggi, H. Zhao and Y. Zhou, (2011) Improving protein fold recognition and template-based modeling by employing probabilistic-based matching between predicted one-dimensional structural properties of the query and corresponding native properties of templates, Bioinformatics 27, 2076-2082 (2011).
Yang, Y.,J. Gao, J. Wang, R. Heffernan, J. Hanson, K. Paliwal and Y. Zhou, (2018). Sixty-five years of the long march in protein secondary structure prediction: the final stretch?“, Briefings in Bioinformatics, 19, 482–494.
Zhou, H. and Y. Zhou, (2004),Single-body residue-level knowledge-based energy score combined with sequence-profile and secondary structure information for fold recognition, Proteins, 55, 1005-1013。
Zhou, H. and Y. Zhou, (2005A), Fold recognition by combining sequence profiles derived from evolution and from depth-dependent structural alignment of fragments, Proteins. 58, 321-328.
Zhou, H. and Y. Zhou, (2005B), SPARKS 2 and SP3 servers in CASP 6.”, Proteins (Supplement CASP issue), Suppl 7 152-156.
Zhou, Y.,Y. Duan, Y. Yang, E. Faraggi, H. Lei, (2011). Trends in template/fragment-free protein structure prediction, Theor. Chem. Accounts 128, 3-16.
https://blog.sciencenet.cn/blog-472757-1348084.html
上一篇:抛弃“影响”因子,计算颠覆因子!
下一篇:强买强卖的中国移动