博文
scCancer2: 基于机器学习整合多篇文献细胞亚群定义的肿瘤微环境深度注释工具
|
王婆卖瓜自卖自夸:scCancer2肿瘤单细胞转录组一站式、微环境深度注释软件包,可以在短时间内获得高准确度、信息量丰富的注释。强烈推荐用于肿瘤单细胞转录组首轮分析,用过都说好!顺带整合肿瘤空间转录组分析功能。
scCancer2软件下载:http://lifeome.net/software/sccancer2/
====================================
scCancer2: 基于机器学习整合多篇文献细胞亚群定义的肿瘤微环境深度注释工具
肿瘤的生存和发展依赖于肿瘤微环境,在细胞水平理解肿瘤微环境的组成对研究肿瘤的发生发展机制有重要意义。近年来,单细胞和空间组学技术迅速发展,极大地促进了肿瘤细胞图谱研究。大量专家标注的肿瘤微环境组学数据得以积累,这为自动化、精准化解析肿瘤微环境特征提供了数据基础。通过数据驱动方法将细胞图谱的标注信息用于肿瘤微环境的注释能为相关研究提供可靠的参考。不同数据集的标注体系存在较大差异,这也为计算分析带来了挑战。
针对以上问题,课题组开发了肿瘤微环境深度注释工具scCancer2 (Chen et al. 2024) ,于2024年1月18日以Original Paper形式在线发表于Bioinformatics期刊。其前身scCancer (Guo et al. 2021)是团队开发的用于肿瘤单细胞转录组一站式分析工具。scCancer集成了常规的单细胞转录组数据分析流程,并重点考虑了肿瘤的样本特征和相关问题,开发了特定的功能模块。例如,全面的数据质量控制与统计分析、肿瘤微环境细胞类型注释、基于inferCNV (Patel et al. 2014)的恶性细胞识别、肿瘤内部异质性的多尺度解析、考虑了肿瘤间异质性的批次效应矫正等。
scCancer2聚焦肿瘤微环境,结合近年来单细胞数据的积累和空间组学技术的发展,从细胞亚型注释、恶性细胞识别和微环境空间特征分析三个方面对scCancer进行升级(图1)。
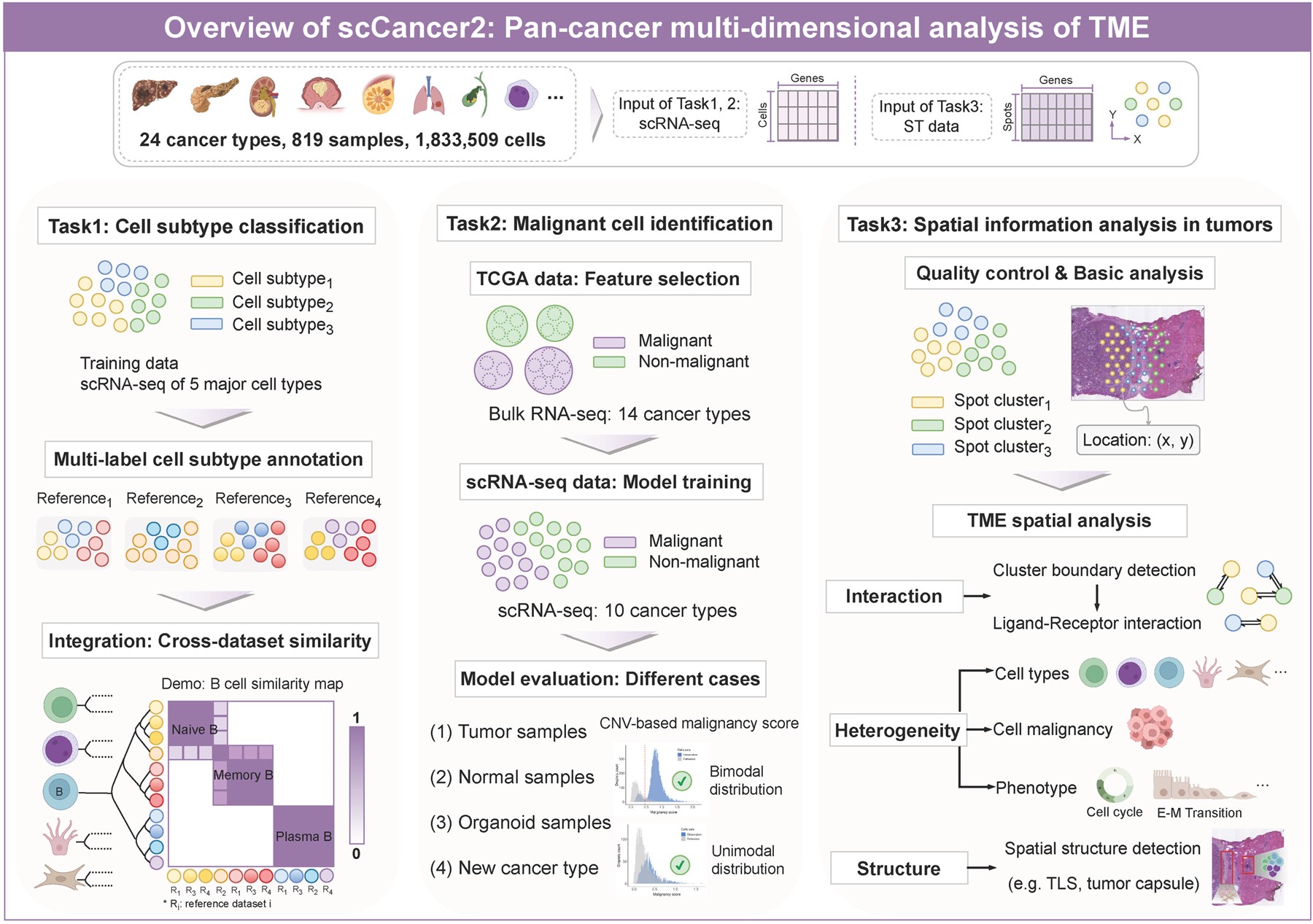
图1:scCancer2肿瘤微环境深度注释的功能模块
在细胞亚型注释方面,目前已有大量精细到细胞亚型水平的公开图谱数据,因此可以通过有监督的机器学习方法训练分类器,注释新的数据集。scCancer2首先考虑提升scCancer肿瘤微环境细胞类型注释的精度,从主要细胞类型精确到细胞亚型水平 (图1,Task1)。相比其他工作,scCancer2聚焦微环境,并整合了大量预先训练的轻量级注释模型。其次,不同的参考数据集之间由于个体异质性、癌种差异、不同专家的标注体系不同等因素,存在显著的差异。在不同的研究中,对于相同的主要细胞类型,有不同的方法或命名标准来定义细胞亚型。为了更好地理解不同文献中标注信息的相似性与差异性,scCancer2通过跨数据集注释定量评估了不同研究中所定义的细胞亚型之间的关系,构建了标签相似性关系图 (图1,Task1)。相似性图提取并归纳了多个图谱数据中的丰富信息,是重要的标注参考和文献索引工具。
在恶性细胞识别方面,scCancer通过改进inferCNV方法实现了对肿瘤样本中细胞恶性程度的评分,通过识别恶性得分的双峰分布来标注恶性细胞。但此类方法仍存在局限性,在样本分布不平衡(例如,缺少内部参考集)的情况下,基于inferCNV的方法性能不稳定。为了更快速、准确地注释恶性细胞,scCancer2整合了多癌种的群体细胞转录组(bulk RNA-seq)和单细胞转录组数据作为参考集,训练了识别恶性细胞的分类器 (图1,Task2)。分类器在多种场景(肿瘤样本、正常样本、肿瘤类器官样本、跨癌种识别)均能给出可靠的分类结果,是对基于inferCNV的注释方法的有效补充。
在微环境空间特征分析方面,scCancer2首先集成了空间转录组基础分析流程,并进一步从三个角度系统地、自动化地解析了肿瘤微环境的空间特征 (图1,Task3):(1)空间相互作用分析:自动识别类别边界并分析边界两侧的相互作用对;(2)空间异质性分析:统计分析肿瘤相关特征在样本不同区域的差异;(3)空间结构识别:开发了基于莫兰指数识别空间结构(三级淋巴结构、肿瘤包膜)的算法,并解析空间结构的细胞组成。
scCancer2在24个癌种的819个样本中进行了模型训练与充分的性能测试,表现出了良好的跨癌种泛化性能。scCancer2的安装与使用指南可从http://lifeome.net/software/sccancer2/中获取。scCancer2的新功能已被集成至scCancer完整分析流程,用户可一站式、自动化地完成全部分析,并从对应文件夹和HTML报告中获取结果。此外,三个功能模块均提供了单独的接口,可以独立调用。
该研究充分利用了现有的大规模肿瘤单细胞及空间转录组学数据,实现了对肿瘤微环境快速、全面、鲁棒的注释,能够为细胞图谱相关研究提供可靠的参考,也具有很高的临床应用价值。
scCancer2软件下载
http://lifeome.net/software/sccancer2/
参考文献
[1] Zeyu Chen, Yuxin Miao, Zhiyuan Tan, Qifan Hu, Yanhong Wu, Xinqi Li, Wenbo Guo, Jin Gu, scCancer2: data-driven in-depth annotations of the tumor microenvironment at single-level resolution, Bioinformatics, 2024, 40(2):btae028, https://doi.org/10.1093/bioinformatics/btae028
[2] Wenbo Guo, Dongfang Wang, Shicheng Wang, Yiran Shan, Changyi Liu, Jin Gu, scCancer: a package for automated processing of single-cell RNA-seq data in cancer, Briefings in Bioinformatics, 2021, 22(3):bbaa127, https://doi.org/10.1093/bib/bbaa127
[3] Patel AP et al., Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science, 2014, 344, 1396-1401, https://doi.org/10.1126/science.1254257
https://blog.sciencenet.cn/blog-407531-1422161.html
上一篇:JGG|区域性表达的基因:基于图的单细胞特征基因选择方法