博文
【R高级教程】专题一:表达谱芯片的聚类分析
|||

【在国际上,R软件的应用是数据分析的主流发展趋势之一,但我发现在国内R软件的使用远不如SPSS、SAS等软件那么流行。为推广R软件的使用,本博客将陆续推出“R高级教程”系列专辑,希望对生命科学领域的科技工作者有少许帮助......】
通常来讲,对于一般的统计分析,基于傻瓜式操作的SPSS(PASW)软件已经足够,但在涉及个性化要求很高的复杂数据处理时,SPSS就开始显得力不从心,这时必须依赖功能更为强大的SAS等软件。以前在自己的科研过程中分析数据多用SPSS、SAS等。在统计遗传和基因组学领域,SAS可以处理很多问题,但与此同时,SAS实现复杂问题过于麻烦,很多问题SAS也不是首选。后来开始运用R环境中的各种免费统计包,特别是Bioconductor的系列分析包,我发觉非常适合生命科学领域的研究者。R有很多优点:
(1)免费,不需要去寻找破解版,不用担心版权问题,使用非常方便;
(2)功能非常强大,单个包的功能比较有限,但多个包组合起来使用则功能无比强大,远胜于SPSS、SAS等;
(3)源代码开放,稍作修改后就能满足个性化的复杂统计分析,满足个性化需求是R的最大特点之一;
(4)程序阅读容易,再加上参考学习资料很多,上手比较容易,提高也不是很难,根据个人经验,要比SAS高级阶段的进阶容易许多;
(5)国际同行高度认同R,我发现很多专用软件都开发了软件的R版,今后R将是数据分析的主流发展方向。
R软件的安装、基本使用等初级教程就不谈了,随便在官方网站找个学习资料就搞定了。“R系列”专辑拟推出中级、高级分析教程。今天推出基因表达谱芯片的聚类分析专题。
本专题示例芯片数据来自GEO数据库中检索号为GSE11787 的Affymetrix芯片的CEL文件,共6个CEL文件,3个正常对照组,3个HPS刺激组,为免疫器官脾脏的表达数据。
(一)原始数据的读入、RNA降解评估和标准化
> pd <- read.AnnotatedDataFrame("Target.txt",header=TRUE,row.names=1,as.is=TRUE)
>rawAffyData <- ReadAffy(filenames=pData(pd)$FileName, phenoData=pd)
> summary(exprs(rawAffyData))
> deg <- AffyRNAdeg(rawAffyData)
> plotAffyRNAdeg(deg, col=c(1,2,3,4,5,6))

> eset <- rma(rawAffyData)
> summary(exprs(eset))
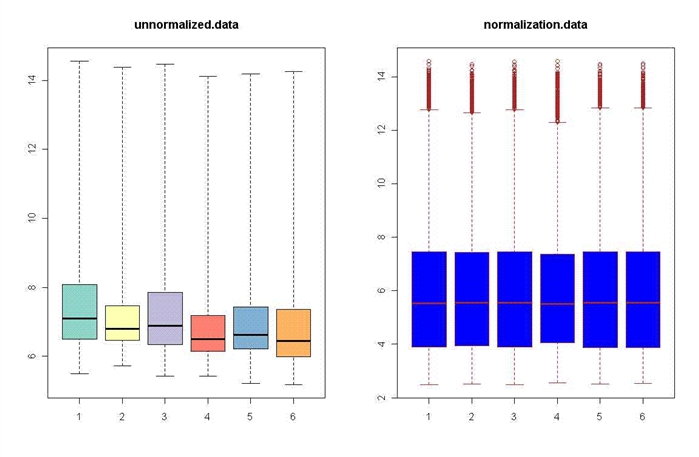
> op <- par(mfrow=c(1,2))
>cols <- brewer.pal(6, "Set3")
>boxplot(rawAffyData,col=cols,names=1:6, main = "unnormalized.data")
>boxplot(data.frame(exprs(eset)) ,names=1:6, main = "normalization.data", col="blue", border="brown")
>par(op)
(二)聚类分析
原始数据读入,经AffyBatch目标转成ExpressionSet目标后,为提高后续分析(如差异表达基因的检测)的统计功效,往往需要进一步经过Detection Call Filter和IQR filter等过滤(“基因芯片数据的特异性过滤与非特异性过滤”将在另一专题里专门讨论)。
需要说明的是,常规做法是先筛选出差异表达基因,然后只用差异表达基因进行聚类分析(本示例直接用了过滤后的数据集,聚类图的效果稍差一点)。
(1)样本聚类
>dd <- dist2(log2(exprs(eset2)))
>diag(dd) <- 0
>dd.row <- as.dendrogram(hclust(as.dist(dd)))
>row.ord <- order.dendrogram(dd.row)
>library("latticeExtra")
>legend <- list(top = list(fun = dendrogramGrob,
args = list(x = dd.row, side = "top")))
>lp <- levelplot(dd[row.ord, row.ord],
scales = list(x = list(rot = 90)),
xlab = "", ylab = "", legend = legend)
>plot(lp)

(2)二维聚类
>source("http://faculty.ucr.edu/~tgirke/Documents/R_BioCond/My_R_Scripts/my.colorFct.R")
>mydata<-exprs(eset2)
>mydatascale <- t(scale(t(mydata)))
>hr <- hclust(as.dist(1-cor(t(mydatascale), method="pearson")), method="complete")
>hc <- hclust(as.dist(1-cor(mydatascale, method="spearman")), method="complete")
>heatmap.2(mydata, Rowv=as.dendrogram(hr), Colv=as.dendrogram(hc), col=redgreen(75), scale="row", ColSideColors=heat.colors(length(hc$labels)), RowSideColors=heat.colors(length(hr$labels)), trace="none", key=T)
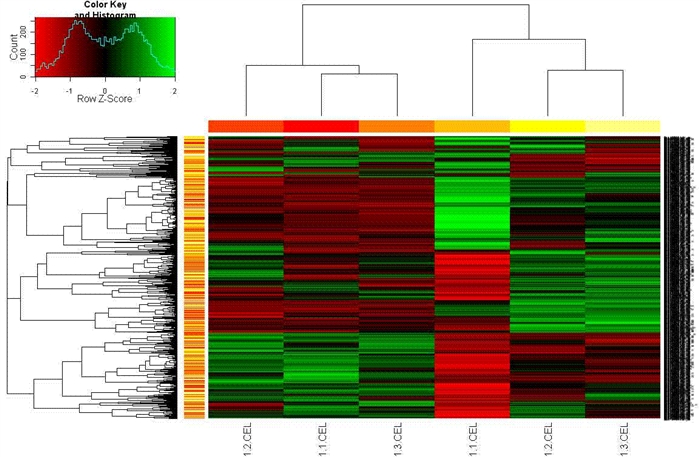
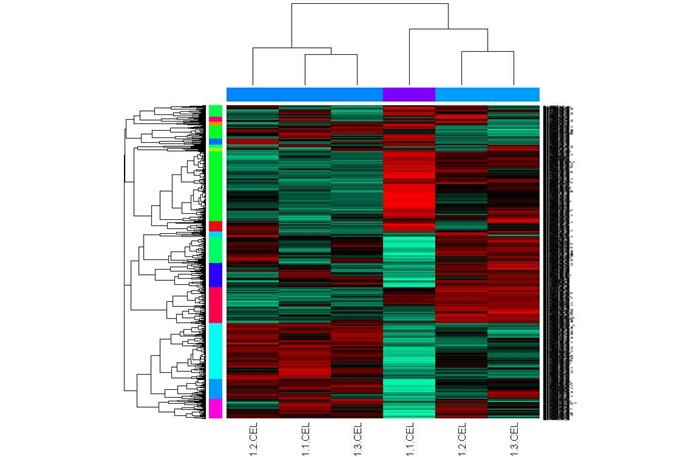
上述聚类图一般和论文里的聚类图有点不同,聚类的模式不太直观,你也可以用下面的语句进行更直观的作图:
>mycl <- cutree(hr, h=max(hr$height)/1.5);
>mycolhc <- sample(rainbow(256)); mycolhc <- mycolhc[as.vector(mycl)]
>myc2 <- cutree(hc, h=max(hc$height)/1.5); mycolhr <- sample(rainbow(256)); mycolhr <- mycolhr[as.vector(myc2)]
>heatmap(mydatascale, Rowv=as.dendrogram(hr), Colv=as.dendrogram(hc), col=my.colorFct(), scale="row", ColSideColors=mycolhr, RowSideColors=mycolhc)
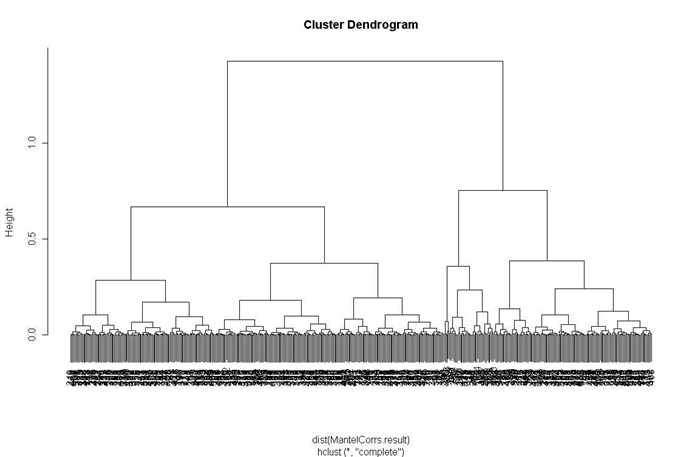
(3)MantelCorrs聚类程序
>kmeans.result <- GetClusters(eset2, 500, 100)
>x=exprs(eset2)
>DistMatrices.result <- DistMatrices(x, kmeans.result$clusters)
>MantelCorrs.result <- MantelCorrs(DistMatrices.result$Dfull,DistMatrices.result$Dsubsets)
>permuted.pval <- PermutationTest(DistMatrices.result$Dfull, DistMatrices.result$Dsubsets,100, 16, 0.05)
>ClusterLists <- ClusterList(permuted.pval, kmeans.result$cluster.sizes,MantelCorrs.result)
>ClusterGenes <- ClusterGeneList(kmeans.result$clusters, ClusterLists$SignificantClusters,eset2)
>h=hclust(dist(MantelCorrs.result))
>plot(h)
【注:除Bioconductor图标外,所有图片均为软件实际运行所得】
https://blog.sciencenet.cn/blog-295006-394794.html
上一篇:本科生科技创新的地位与作用
下一篇:也谈中国科研问题的根源