博文
氢气作为抗氧化剂与辐射防护剂:来自蒙特卡罗辐射化学模拟的机制见解
||
氢气作为抗氧化剂与辐射防护剂:来自蒙特卡罗辐射化学模拟的机制见解
作者:苏迈亚·阿克特尔·里亚(Sumaiya Akhter Ria)、金塔纳·米森诺恩(Jintana Meesungnoen)、让-保罗·杰林(Jean-Paul Jay-Gerin)*(ORCID编号:略)
加拿大魁北克省舍布鲁克市12号大道北3001号,舍布鲁克大学医学与健康科学学院医学影像与放射科学系,邮编:J1H 5N4
Ria, S.A.; Meesungnoen, J.; Jay-Gerin, J.-P. Molecular Hydrogen as an Antioxidant and Radioprotector: Mechanistic Insights from Monte Carlo Radiation-Chemical Simulations. Antioxidants 2025, 14, 1054.
摘要
(1)背景:水约占细胞质量的70%-80%,是活细胞中含量最丰富的成分。当暴露于电离辐射时,水会发生辐射分解,产生包括自由基和分子产物在内的多种活性物质。其中,羟基自由基(•OH)因其极高的反应活性以及对DNA、膜脂质、蛋白质等重要生物分子的氧化损伤能力,具有极强的破坏性。本研究从辐射化学视角出发,探究氢分子(H₂)对•OH自由基的选择性清除能力,旨在评估其作为抗氧化剂和辐射防护剂的潜力;(2)方法:采用蒙特卡罗径迹化学模拟程序IONLYS-IRT,对中性有氧水环境中活性氧(ROS)的时间依赖性产额进行建模。模拟中设置了不同浓度的溶解态H₂,并以已知含硫辐射防护剂与抗氧化剂——胱胺(cystamine)作为对照。辐射源选用300 MeV质子,以模拟低线性能量转移(LET)辐射(如⁶⁰Co γ射线或快电子(>1 MeV))的辐射分解效应;(3)结果:模拟结果定量表明,H₂可选择性清除•OH自由基。然而,其清除效率始终低于胱胺:由于胱胺反应活性更高、自由基猝灭能力更强,对•OH的抑制作用更快且更显著;(4)结论:氢分子具有多项独特优势,包括低毒性、高扩散性、对•OH自由基的选择性清除能力,以及已被充分证实的抗炎作用。尽管在自由基清除效率方面不及胱胺,但其优异的安全性和生物相容性使H₂成为一种极具潜力的辐射防护剂与抗氧化剂,可用于针对辐射诱导氧化应激与炎症的治疗应用。
1. 引言
人体主要由水构成,水在体温调节、营养运输、废物排出、细胞代谢等关键生理功能中发挥核心作用。当暴露于电离辐射时,辐射与细胞内水分子发生主要相互作用,导致水分子电离与电子激发[1,2]。电离过程产生极不稳定的水自由基阳离子(H₂O•⁺),其会在约50飞秒(fs)内[3],通过与相邻水分子发生准一级质子转移反应快速分解,生成羟基自由基(•OH)和水合氢离子(H₃O⁺),反应如下:
H₂O•⁺ + H₂O → H₃O⁺ + •OH (1)
与此同时,激发过程产生电子激发态水分子(H₂O*),其在液相中主要通过解离生成•OH自由基和氢原子(H•),反应为:H₂O* → H• + •OH[4]。尽管激发过程会促成自由基形成,但其总体影响相较于主导的电离驱动过程仍较微弱。
羟基自由基是最强的氧化剂之一,可与核酸、脂质、蛋白质发生无差别反应。据估算,在哺乳动物细胞中,X射线或γ射线诱导的DNA损伤约有三分之二由•OH引起[5,6]。例如,在受辐射的生物环境中,•OH可轻易从相邻生物有机分子(RH)中夺取氢原子,反应如下:
•OH + RH → R• + H₂O (2)
该反应的速率常数(k)通常在10⁸-10⁹ M⁻1s⁻1范围内[7]。在氧化应激条件下,生成的碳中心自由基(R•)会迅速与分子氧(O₂)反应,形成过氧自由基(ROO•):
R• + O₂ → ROO• (3)
此反应速率接近或达到扩散控制极限(k≈2×10⁹ M⁻1s⁻1)。这些过氧自由基的氧化能力往往强于其前体自由基。一旦形成,过氧自由基会对母体分子造成不可逆修饰——这一过程被称为分子氧的“损伤固定”作用,即O₂使辐射诱导的初始变化永久化[7,8]。
由于反应活性极高,•OH、过氧自由基及其他被统称为“活性氧(ROS)”的物质可引发氧化链式反应,其中以脂质过氧化和DNA氧化尤为典型。在生理浓度(即低水平)下,ROS也在细胞信号传导和免疫防御中发挥重要作用[9]。然而,当ROS水平超过内源性抗氧化剂的缓冲能力(如放疗中可能出现的情况)时,会引发氧化应激失衡。这种状态会对DNA、蛋白质、膜脂质等关键细胞大分子造成广泛且常为不可逆的损伤,最终损害细胞活力并扰乱正常生理功能[9,10,11]。持续的氧化应激还与多种疾病的发病机制相关,如癌症、心血管疾病、阿尔茨海默病和帕金森病等神经退行性疾病,甚至与生物衰老过程有关[10,12,13]。
水辐射分解的生物学意义还体现在纳米材料辅助放疗中[14]:高原子序数(高Z)或具有催化活性的纳米颗粒可在局部增强•OH及其他ROS的辐射分解产额,从而加剧DNA损伤并提高肿瘤细胞杀伤效率。在此背景下,本研究对氢分子清除•OH的定量分析,为理解抗氧化剂与辐射防护剂如何调控这些过程提供了机制层面的见解,对优化治疗效果具有潜在意义。
为减轻ROS诱导的损伤(无论损伤源于正常生理条件还是电离辐射暴露),细胞依赖复杂的内源性抗氧化防御网络。该网络包括谷胱甘肽、抗坏血酸(维生素C)、α-生育酚(维生素E)等小分子,以及超氧化物歧化酶(SOD)、过氧化氢酶、谷胱甘肽过氧化物酶(一种含硒酶)等酶系统。这些成分通过协同作用清除ROS:或提供电子/氢原子,或催化ROS转化为反应活性更低的物质。
然而,当ROS生成量超过内源性抗氧化防御系统的中和能力时,可能需要补充外源性抗氧化剂以恢复氧化还原平衡。抗氧化剂的细胞保护效能主要取决于其自由基清除能力,即能否在自由基对关键细胞靶点造成氧化损伤前将其中和[10,15]。
从辐射化学视角看,许多抗氧化剂可作为有效的化学(非生物)辐射防护剂。其保护作用源于对细胞内水辐射分解过程中产生的高活性物质(包括•OH、水合电子(e⁻aq)、H•、过氧化氢(H₂O₂),以及有氧条件下的氢过氧自由基(HO₂•)、超氧阴离子自由基(O₂•⁻)等过氧物质[17])的清除能力[1,2,16]。这种ROS清除能力是辐射防护的核心机制,与抗氧化剂对抗氧化损伤的经典作用高度一致。在此背景下,抗氧化活性是辐射防护剂发挥细胞保护作用的主要途径[18,19,20,21,22,23,24]。
值得注意的是,目前尚未发现能清除•OH的酶系统,而•OH被广泛认为是所有ROS中细胞毒性最强的物质。因此,直接清除•OH是抗氧化防御的关键环节。已有研究表明,氢分子(H₂)可通过与羟基自由基特异性反应,发挥选择性、无毒的抗氧化剂或辐射防护剂作用[25,26,27,28,29,30,31,32,33,34],如下列放热反应所示[2,10,35]:
•OH + H₂ → H• + H₂O (4)
在25℃的水中,该反应的速率常数为4×10⁷ M⁻1s⁻1[36]。在有氧细胞环境中,生成的氢原子(H•)会被氧迅速清除,形成HO₂•自由基:
H• + O₂ → HO₂•,k=1.3×101⁰ M⁻1s⁻1 (5)
在生理pH条件下,HO₂•以去质子化形式(O₂•⁻)存在[17],随后在SOD催化下发生歧化反应[37]:
O₂•⁻+ O₂•⁻+ 2H⁺→ O₂+ H₂O₂,k≈4×10⁹ M⁻1s⁻1 (6)
综上,反应(4)-(6)通过将自由基化学过程导向反应活性更低、更易被内源性抗氧化系统中和的物质,从而避免•OH相关的生物分子损伤。
除与•OH反应外,H₂还被证实可减轻过氧亚硝酸盐(ONOO⁻)及其共轭酸过氧亚硝酸(ONOOH,37℃下pKa≈6.8)的损伤效应——这两种高细胞毒性的“活性氮(RNS)”可对多种生物靶点造成损伤[38,39]。在生理条件下,ONOO⁻由一氧化氮(•NO,又称一氧化氮自由基,大多数哺乳动物细胞内源性产生的脂溶性链断裂自由基)与超氧阴离子自由基通过快速的扩散控制反应生成[40,41]:
•NO + O₂•⁻ → ONOO⁻ (7)
该反应的速率常数为1.9×101⁰ M⁻1s⁻1,远高于生理pH条件下SOD催化O₂•⁻歧化反应(反应(6))的速率常数。
然而,迄今为止,H₂与过氧亚硝酸盐的直接反应:
ONOO⁻ + H₂ → NO₂⁻ + H₂O (8)
其机制仍不明确,且未进行动力学表征。尽管如此,实验研究表明,在多种生物系统中,溶解态H₂可减轻ONOO⁻相关的氧化损伤[27]。目前认为,反应(8)并非扩散控制反应,其速率远慢于反应(4)。尽管H₂与ONOO⁻相互作用的机制细节仍需进一步阐明,但现有证据表明,H₂的抗氧化与辐射防护作用可能更为广泛——不仅限于以氧为中心的自由基,还包括辐射或炎症应激过程中产生的含氮氧化剂。
相较于胱胺(一种含硫化合物,反应活性更高,可快速清除自由基[24,42])等传统辐射防护剂或抗氧化剂,氢分子与羟基自由基的反应速率更慢。H₂反应活性相对较低的原因在于其稳定的非极性分子结构。尽管如此,H₂常被认为是优于胱胺的抗氧化剂,因其具有多项独特优势:可选择性中和毒性最强的物质(尤其是•OH和ONOO⁻),同时具备优异的生物相容性和固有无毒性[27,31]。此外,H₂分子体积小、扩散性高,可快速穿过生物膜(包括血脑屏障[43,44,45]),因此能在中枢神经系统内发挥抗氧化作用,有助于预防或延缓神经退行性病变的发生[46,47]。
此外,研究人员已开发出多种便捷高效的H₂体内递送方法。如文献[31,33,34]中详细综述所示,这些方法包括吸入H₂气体、口服富氢水(H₂-water)、静脉或腹腔注射富氢生理盐水(H₂-saline)、通过富氢水浴局部给药,以及使用富氢生理盐水滴眼液眼部给药。除抗氧化和细胞保护特性外,H₂的安全性、易用性及递送方式的多样性显著提升了其作为预防和治疗剂的吸引力,使其比许多传统抗氧化剂或辐射防护剂更易被接受和获取。
本研究的目的是:以300 MeV质子(用于模拟⁶⁰Co γ射线或快电子等传统低线性能量转移(LET)辐射)为辐射源,在室温条件下,探究有无添加H₂时,有氧和无氧水辐射分解过程中辐射分解产物的时间依赖性演变。通过蒙特卡罗径迹化学模拟[4,42,48],重点研究在有无溶解氧的中性水中,H₂是否通过选择性降低羟基自由基产额发挥真正的抗氧化作用;同时探究不同H₂浓度(0-10 mM)对•OH产额的影响——浓度范围不仅涵盖常见实验或临床应用的约0.3 mM,还延伸至标准大气压下饱和富氢饮用水的溶解度极限(约0.78 mM,即1.57 mg/L或1.57 ppm[27,29,49,50]);最后,在相同浓度条件下,评估胱胺清除•OH的抗氧化效率,并与H₂的性能进行比较。
所有模拟均在25℃下进行。辐射化学产额以每吸收100 eV能量产生的分子数表示,其中初级产额用g(X)表示,实验值用G(X)表示。为与国际单位制(SI,单位:mol/J)统一,采用以下换算关系:1分子/100 eV≈0.10364 μmol/J[1,2]。
2. 材料与方法
2.1 纯无氧水与有氧水的低线性能量转移(LET)辐射分解:事件时间尺度、自由基与分子产物的形成及蒙特卡罗径迹化学建模
水的辐射分解是一个复杂的多阶段过程,起始于电离辐射的初始能量沉积(即物理阶段)。随后进入物理化学阶段与空间非均匀化学阶段,在此过程中,活性物质会沿辐射径迹形成并在不断扩展的“射束”(spur)内演变[1,51,52]。在低LET辐射条件下,25℃时这些过程通常在初始电离事件发生后约0.2微秒(μs)内完成[53]。超过此时间点后,射束消散,剩余的辐射分解产物通常被认为在整个本体溶液中呈均匀分布。文献[1]近期已对这些阶段进行了详细阐述。
在⁶⁰Co γ射线、快电子或数百兆电子伏特(MeV)质子(LET≈0.3千电子伏特/微米(keV/μm))作用下,纯无氧水发生辐射分解时,均匀分布状态下的主要活性物质包括“自由基”产物(水合电子e⁻aq、氢原子H•、羟基自由基•OH)与“分子”产物(氢气H₂、过氧化氢H₂O₂)。此时这些物质的产额(传统上称为初级产额或“逃逸”产额)如下[1]:
g(e⁻aq)=2.65,g(H•)=0.60,g(H₂)=0.45,g(•OH)=2.80,g(H₂O₂)=0.68 (9)
值得注意的是,大部分H₂产生于辐射分解的早期物理化学阶段,而非射束内化学反应[54,55,56]。此外,作为气态产物,辐射分解生成的H₂往往易从溶液中逸出;但若被保留在溶液中,其可通过反应(4)与氧化性•OH自由基发生反应。这一情况与本研究高度相关,因为本研究旨在探究富氢水(即添加了H₂的水)的辐射分解过程。
本研究采用实验室自主开发的蒙特卡罗径迹化学程序IONLYS-IRT,模拟25℃下、300 MeV质子辐射作用下,有无添加H₂和胱胺时有氧水与无氧水的辐射分解过程。如前所述,这些质子可模拟⁶⁰Co γ射线或快电子的低LET辐射效应。该程序已通过与全球多个实验室在不同条件(pH、温度、剂量率、LET、溶质组成各异)下获得的实验数据进行广泛验证,充分证明其在多种辐射条件下的稳定性与可靠性。程序的详细说明可参见其他文献(如[4]及其中引用的文献)。
简要来说,我们的逐事件IONLYS程序[57]可模拟辐射作用早期物理阶段与物理化学阶段(约1皮秒(ps)内)发生的所有事件。模拟得到的物质(e⁻aq、H₃O⁺、OH⁻、H•、H₂、•OH、H₂O₂、HO₂•/O₂•⁻、•O•(3P)、O•⁻等)复杂且高度非均匀的空间分布,将作为后续化学阶段模拟的输入数据。在第三阶段(>1 ps),辐射分解产物根据其扩散系数随机扩散,并与其他产物或辐射过程中存在的溶解溶质(如本研究中涉及的有氧溶液中的氧气、H₂或胱胺)发生反应。此阶段采用IRT程序[58]建模,该程序基于“独立反应时间”(IRT)方法[59,60,61]——这是一种计算高效的随机方法,无需追踪单个粒子的轨迹。通过与多种辐射条件下的完整随机飞行(或“逐步”)蒙特卡罗模拟结果对比,已验证该方法的准确性[62,63]。值得注意的是,当辐射径迹消失且辐射分解产物在本体溶液中均匀分布时,IRT程序也可有效用于模拟后期的均相化学反应。
在常规辐射条件下(即无剂量率效应[2,64]),辐射分解产物的浓度相较于溶解氧、H₂和胱胺的背景浓度始终较低。因此,在IRT程序中,这些物质的反应可建模为准一级反应。
IRT程序还考虑了离子强度对所有离子-离子反应的影响,但水合电子e⁻aq的双分子自重组反应除外——目前尚无报道表明该反应与离子强度相关[65]。速率常数的调整采用文献[48,66]中所述的相同方法。
IRT模拟中涉及的各类物质的扩散系数均引自文献[58,67]。25℃时,H₂的扩散系数取值为5.1×10⁻⁵平方厘米/秒(cm2/s);有氧溶液中氧气的扩散系数取值为2.4×10⁻⁵ cm2/s。对于含胱胺的溶液,基于胱胺及其所有辐射分解衍生物的分子大小相似且缺乏各物质的特定扩散数据,胱胺及其所有辐射分解衍生物的扩散系数均采用通用值2×10⁻⁵ cm2/s[48]。
最后,本研究忽略了电离辐射对溶质(O₂、H₂、胱胺)的“直接”作用。这一近似处理的合理性在于:与本体水(≈55.5摩尔/升(M))相比,本研究中溶质的浓度较低(O₂为0.25 mM,H₂或胱胺为0-10 mM)。
所有计算均通过模拟300 MeV质子的短径迹片段(通常为50-150微米(μm))完成。25℃时,模拟得到的平均LET值约为0.3 keV/μm,该值与Watt[68]报道的数据及国际辐射单位与测量委员会(ICRU)第49号报告[69]中针对密度为1克/立方厘米(g/cm3)的液态水推荐的LET值高度一致。因此,这些模型计算得出的“径迹片段”产额对应明确的LET值。模拟质子事件数(通常为40-100个)的选择标准为:在保证合理计算时间的同时,使平均化学产额的统计不确定性最小化。
2.2 溶解氧在水辐射分解中的作用
25℃下空气饱和的水中(溶解氧浓度约为0.25 mM),O₂与e⁻aq和H•的反应如下[1]:
e⁻aq + O₂ → O₂•⁻,k=2.3×101⁰ M⁻1s⁻1 (10)
H• + O₂ → HO₂•,k=1.3×101⁰ M⁻1s⁻1 (5)
其中,O₂•⁻与其共轭酸HO₂•处于pH依赖性平衡状态(见上文)。23℃时,HO₂•/O₂•⁻对的解离常数(pKa)为4.8[17]。根据该pKa值,通过亨德森-哈塞尔巴尔赫方程可推断:在中性有氧水或生理pH条件下的有氧细胞环境中,O₂•⁻是氢过氧自由基的主要存在形式。
在有氧水中,O₂清除e⁻aq或H•的时间尺度约为0.2-0.4 μs,该值通过“清除能力”的倒数估算得出——“清除能力”即溶质浓度与其与初级自由基反应的速率常数的乘积[2,70]。此时间尺度大致对应低LET辐射条件下射束扩展的结束时间与均相化学反应的起始时间。
2.3 含胱胺条件下水辐射分解的建模:反应方案
胱胺是一种有机二氨基二硫化物化合物,分子式为RSSR(其中R=NH₂–CH₂–CH₂),是半胱胺(RSH)的二硫化物形式。半胱胺是一种氨基硫醇,源自半胱氨酸(HS–CH₂–CH(NH₂)–COOH)——一种存在于大多数蛋白质中的关键氨基酸。自Bacq及其同事开展开创性研究以来[24,71,72],胱胺的辐射防护特性已为人熟知;其在减轻细胞和组织内氧化应激、保护细胞免受辐射诱导损伤方面发挥关键作用。
在模拟含胱胺有氧水的辐射分解过程时,IRT程序中采用的化学反应方案、速率常数及活性物质扩散系数,均基于此前对弗里克-胱胺溶液中亚铁离子辐射氧化为铁离子的研究[24,42,48]。值得注意的是,我们的胱胺辐射化学模型无需调整参数,即可准确再现⁶⁰Co γ射线或快电子辐射下硫酸亚铁(弗里克)-胱胺水溶液中Fe3⁺的实验产额[42]。且在广泛的胱胺浓度范围内(无论有无氧气),该模型的准确性均保持一致[42]。模拟得出的G(Fe3⁺)值与观测数据的高度吻合,验证了该反应方案的可靠性。本研究中,水辐射分解反应方案(详见[4]的表14.1)新增了17个化学反应[24],具体列于表1。
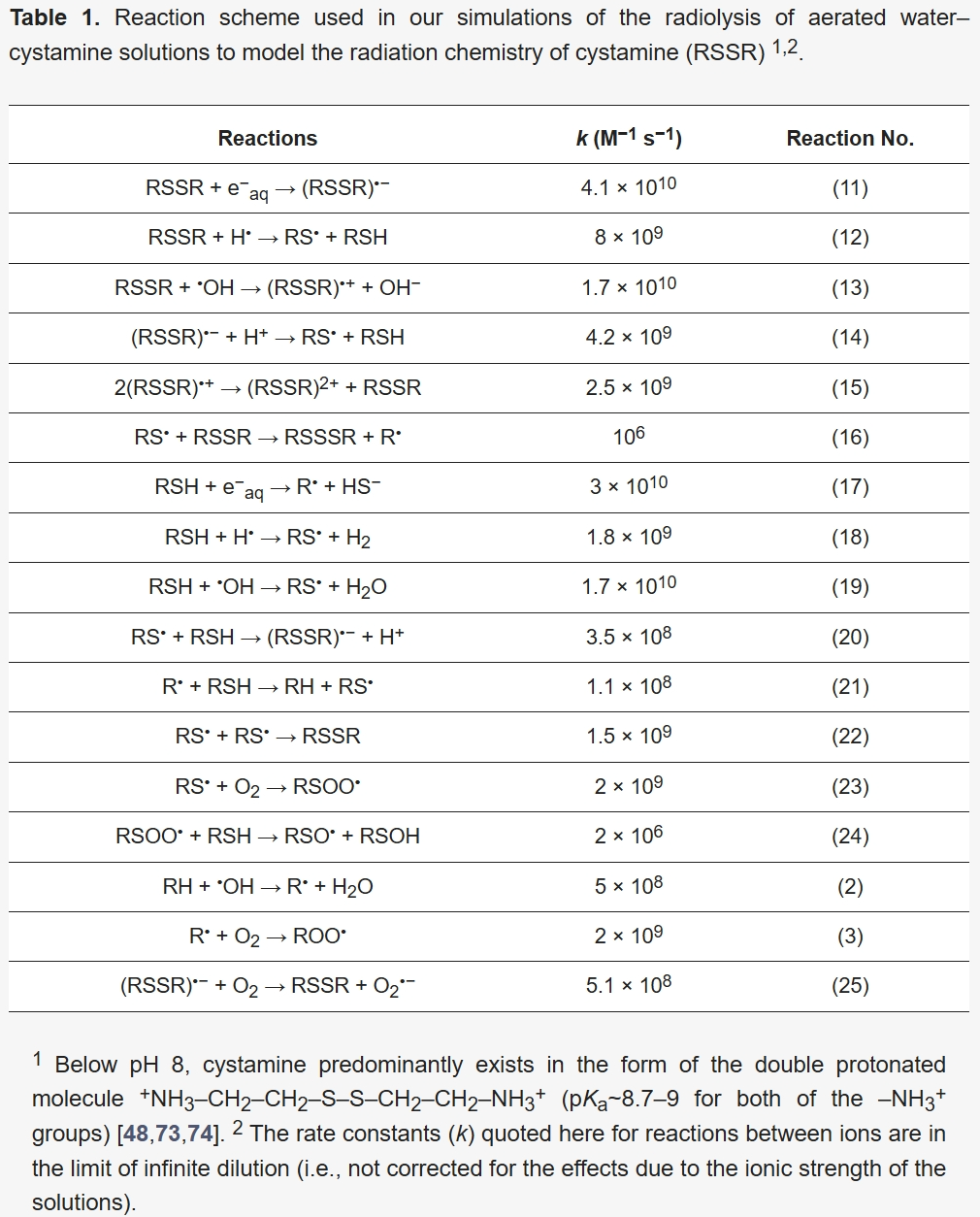
表1 模拟含胱胺有氧水辐射分解时采用的反应方案(用于模拟胱胺(RSSR)的辐射化学)1·2
3. 结果
3.1 无氧条件下有无添加H₂的水溶液辐射分解中活性物质的产额
图1a、b对比了25℃下、300 MeV质子(LET≈0.3 keV/μm)辐射作用下,无氧中性水在未添加H₂和添加1 mM H₂时,1 ps至10毫秒(ms)内辐射分解产物产额的时间演变。结果清晰表明,添加H₂会显著改变•OH和H•的产额,而其他辐射分解产物的产额基本保持不变。
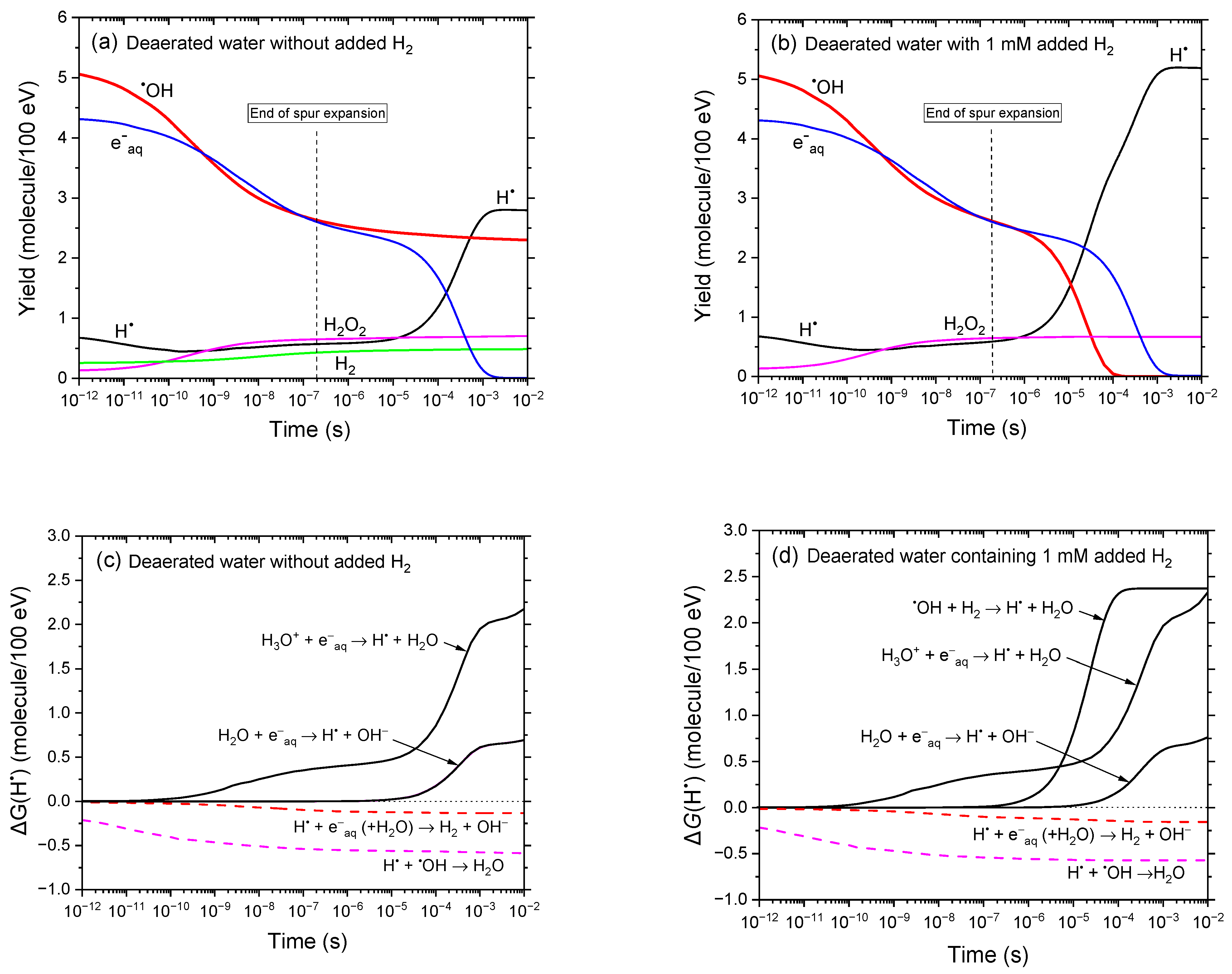
图1 基于IONLYS-IRT蒙特卡罗径迹化学模拟的25℃下300 MeV质子(LET≈0.3 keV/μm)辐射无氧水时,关键活性物质(e⁻aq、•OH、H•、H₂O₂、H₂)产额在1 ps至10 ms内的时间演变。(a)(b)分别为未添加H₂和添加1 mM H₂时的结果。作为参考,约0.2 μs处的垂直虚线表示溶液从非均匀射束动力学向本体均相动力学的转变[53]。(c)(d)分别为未添加H₂和添加H₂时,由相同蒙特卡罗模拟计算得出的各反应对H•形成(黑色实线)和衰减(红色虚线、品红色虚线)的时间依赖性贡献∆G(H•)(详见正文)。**
添加H₂后,在1-100 μs内观察到•OH的产额G(•OH)显著降低(约减少2.5个G单位)(图1b),这与反应(4)一致,表明氢分子具有强大的•OH清除能力。G(•OH)持续下降,至约100 μs时接近零。相反,无论有无添加H₂,无氧水辐射分解都会生成H•自由基;但如图1a、b所示,添加H₂后G(H•)显著增加:10 ms时,未添加H₂的G(H•)约为2.8分子/100电子伏特(molecules/100 eV),而添加1 mM H₂后增至约5.2 molecules/100 eV。图1c、d进一步阐释了这一现象——该图展示了300 MeV质子辐射模拟中,各反应对H•形成与衰减的时间依赖性贡献∆G(H•)。未添加H₂时,H•的主要来源是以下反应[4,36]:
H₃O⁺ + e⁻aq → H• + H₂O,k=2.1×101⁰ M⁻1s⁻1 (26)
H₂O + e⁻aq → H• + OH⁻,k=15.8 M⁻1s⁻1 (27)
当存在溶解态H₂时,反应(4)会产生额外的显著贡献——在该反应中,•OH被H₂还原为H•,进一步提高了G(H•)的总产额。
从放射生物学角度来看,尽管H•的反应活性和破坏性低于•OH,但它仍是一种具有化学活性的自由基,绝非生物学惰性物质[7,10]。这在缺氧环境(如实体瘤中常见的环境)中可能具有重要意义:缺氧条件下,氧气的缺失会限制本可中和H•的后续反应(如反应(5)和(6))。在此类环境中,H•自由基的积累可能引发次级化学过程(包括生物分子的还原或与金属离子的相互作用),进而可能影响治疗效果。因此,尽管H₂具有抗氧化作用,但其在缺氧组织中的辐射防护作用可能比单纯“消除”氧化应激更为复杂。
3.2 有氧条件下有无添加H₂的水溶液辐射分解中活性物质的产额
图2a、b对比了25℃下、300 MeV质子辐射作用下,有氧中性水在未添加H₂和添加1 mM H₂时,1 ps至10 ms内辐射分解产物产额的时间演变。与图1a、b类似,添加H₂会显著改变•OH和H•的产额;但在此情况下,溶解氧的同时存在会进一步改变体系——将e⁻aq转化为O₂•⁻,将H•转化为HO₂•。图2c、d进一步阐释了这些效应,展示了蒙特卡罗模拟计算得出的未添加H₂和添加1 mM H₂时,各反应对H•形成与衰减的时间依赖性贡献∆G(H•)。作为对比,图1c、d呈现了无氧条件下的相应结果。
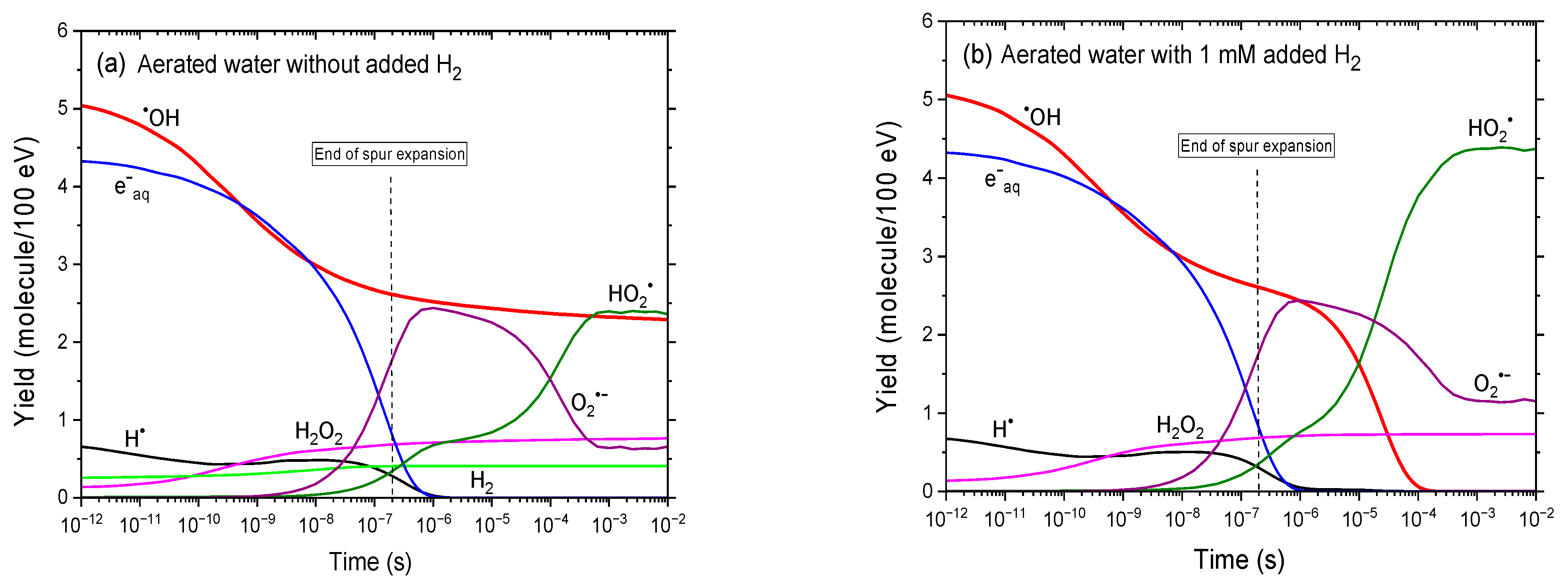
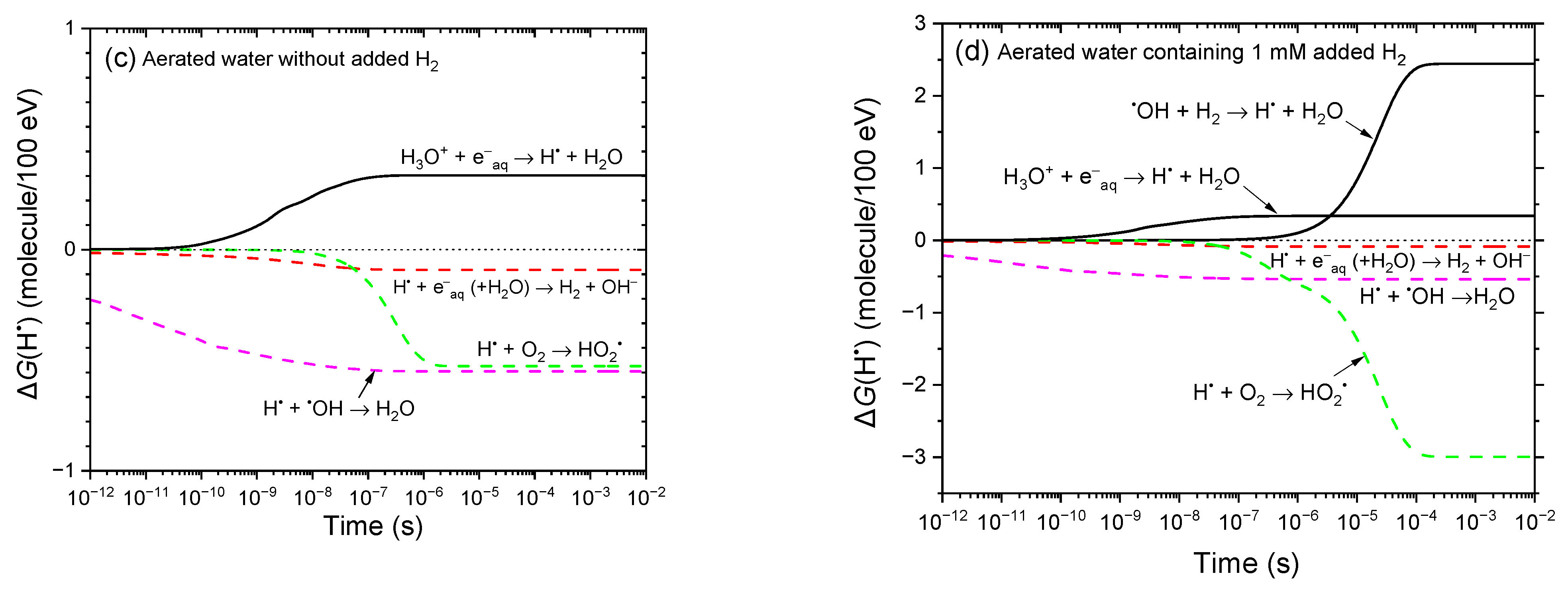
图2 基于IONLYS-IRT蒙特卡罗径迹化学模拟的25℃下300 MeV质子(LET≈0.3 keV/μm)辐射有氧水时,关键活性物质(e⁻aq、•OH、H•、H₂O₂、H₂、O₂•⁻、HO₂•)产额在1 ps至10 ms内的时间演变。计算中采用的溶解氧浓度为0.25 mM。(a)(b)分别为未添加H₂和添加1 mM H₂时的结果。作为参考,约0.2 μs处的垂直虚线表示溶液从非均匀射束动力学向本体均相动力学的转变[53]。(c)(d)分别为未添加H₂和添加H₂时,由相同蒙特卡罗模拟计算得出的各反应对H•形成(黑色实线)和衰减(绿色虚线、红色虚线、品红色虚线)的时间依赖性贡献∆G(H•)(详见正文)。
下文将对这些图进行详细分析。
未添加氢气(H₂)时,羟基自由基产额(G(•OH))的时间演变与无氧条件下基本一致。然而,在溶解氧浓度为0.25 mM的体系中,根据氧气清除能力的倒数估算,氢原子(H•)会在约0.3微秒(μs)时通过反应(5)转化为氢过氧自由基(HO₂•)。此外,与无氧水辐射分解不同,水合电子(e⁻aq)会通过反应(10)被氧气快速清除,并在约0.2 μs时形成超氧阴离子自由基(O₂•⁻)。因此,几乎所有的e⁻aq都会有效转化为O₂•⁻,这也解释了为何在约10纳秒(ns)至1 μs内观察到G(O₂•⁻)急剧上升。在更长的时间尺度上,由于以下反应[4,36],G(O₂•⁻)会逐渐下降:
O₂•⁻ + H₃O⁺ → HO₂• + H₂O,速率常数k = 5×101⁰ M⁻1s⁻1 (28)
这种下降趋势会持续到约1毫秒(ms)时,G(O₂•⁻)稳定在约0.66分子/100电子伏特(molecules/100 eV)。因此,HO₂•的产额(G(HO₂•))主要来源于两部分:一是H•与O₂的直接反应(反应(5)),二是O₂•⁻的质子化反应(反应(28))。如图2a所示,G(HO₂•)同样在1 ms左右趋于稳定,最终达到约2.36 molecules/100 eV。由此可知,在毫秒时间尺度内,HO₂•与O₂•⁻的产额之和约为3.02 molecules/100 eV。作为对比,在未添加H₂的无氧水辐射分解体系中,同一时间点的H•产额(G(H•))达到约2.80 molecules/100 eV(图1a)。
综上,在溶解氧浓度为0.25 mM且未添加H₂的有氧水中,约1 ms时体系中剩余的氧化性物质仅有•OH、HO₂•/O₂•⁻和过氧化氢(H₂O₂),其产额分别为约2.3、3.02和0.76 molecules/100 eV。
除上述氧气带来的影响外,向有氧水中添加1 mM H₂会导致•OH在约100 μs后完全消失,同时通过反应(4)生成额外的H•,随后这些H•又会通过反应(5)转化为HO₂•。因此,在更长时间尺度下,体系中剩余的自由基物种仅有HO₂•/O₂•⁻。约10 ms时,二者的产额之和达到约5.52 molecules/100 eV,这一数值与添加1 mM H₂的无氧水辐射分解体系中H•的产额(约5.2 molecules/100 eV,图1b)相当。而H₂O₂的产额则相对稳定,不受氧气或H₂存在与否的影响。
如前所述,在25℃的中性有氧水或生理pH值的有氧细胞环境中,O₂•⁻是氢过氧自由基的主要存在形式[10,17]。也就是说,从放射生物学角度来看,这种从高活性•OH向活性较低但寿命更长的O₂•⁻的转化,可能会显著改变辐射诱导损伤的性质:一方面可能减少对DNA和蛋白质的直接氧化损伤,另一方面则更易引发缓慢的氧化还原驱动反应[7,10]。不过,这些O₂•⁻最终应会被内源性抗氧化系统中和,尤其是通过超氧化物歧化酶(SOD)催化的歧化反应(6)。
尽管如此,这些分析表明,虽然H₂能缓解氧化应激,但其整体放射生物学效应(尤其是在缺氧组织中)可能比预期的更为复杂。
3.3 不同H₂浓度下有氧水辐射分解中G(•OH)的时间变化曲线
图3a展示了在300 MeV质子辐射作用下,添加不同浓度H₂(0.01-10 mM)的有氧水辐射分解过程中,G(•OH)随时间的演变。结果显示,随着H₂浓度的升高,•OH产额开始下降的时间逐渐提前。下降起始点(定义为G(•OH)曲线偏离未添加H₂曲线的时刻)从0.01 mM H₂时的约60 μs提前至10 mM H₂时的约30 ns。这一现象反映出清除反应(4)的效率增强——在此反应中,•OH被H₂消耗(见上文)。在所有研究的H₂浓度下,几乎所有辐射分解产生的•OH最终都会通过该反应被清除。
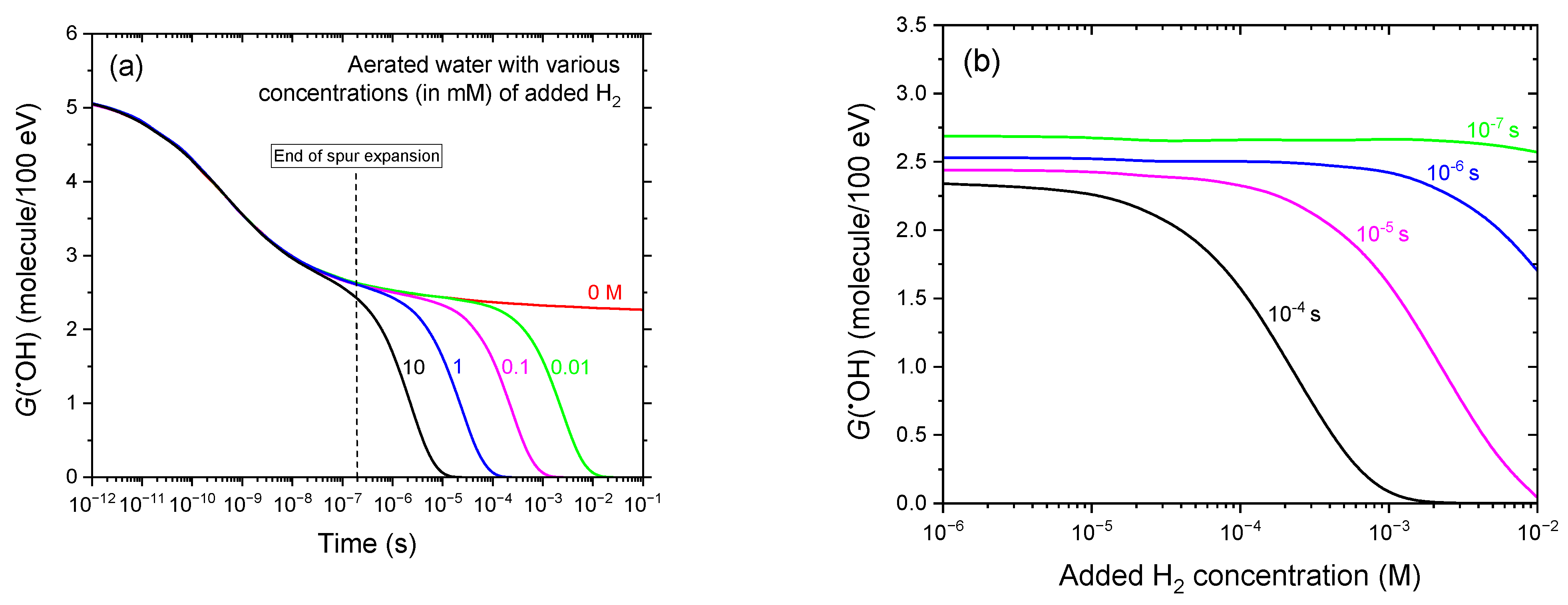
图3 (a)基于IONLYS-IRT蒙特卡罗径迹化学模拟的25℃下300 MeV质子(LET≈0.3 keV/μm)辐射有氧水时,不同H₂浓度(单位:mM)下•OH产额随时间(1 ps-100 ms)的演变:10(黑色曲线)、1(蓝色)、0.1(品红色)、0.01(绿色)。红色曲线代表未添加H₂时的G(•OH)。10 mM H₂的曲线仅用于与10 mM溶解态胱胺的对应结果(图4)进行对比。需注意,H₂在水中的溶解度较低——标准环境温度和压力下约为0.78 mM(1.57 mg/L或1.57 ppm)(见正文)。作为参考,约0.2 μs处的垂直虚线表示溶液从非均匀射束动力学向本体均相动力学的转变[53]。(b)为图3a的另一种呈现形式,展示了蒙特卡罗模拟得出的辐射后不同时间点(10⁻⁷、10⁻⁶、10⁻⁵、10⁻⁴ s)下,G(•OH)随H₂浓度(10⁻3-10 mM)升高的下降趋势。计算中采用的溶解氧浓度为0.25 mM。
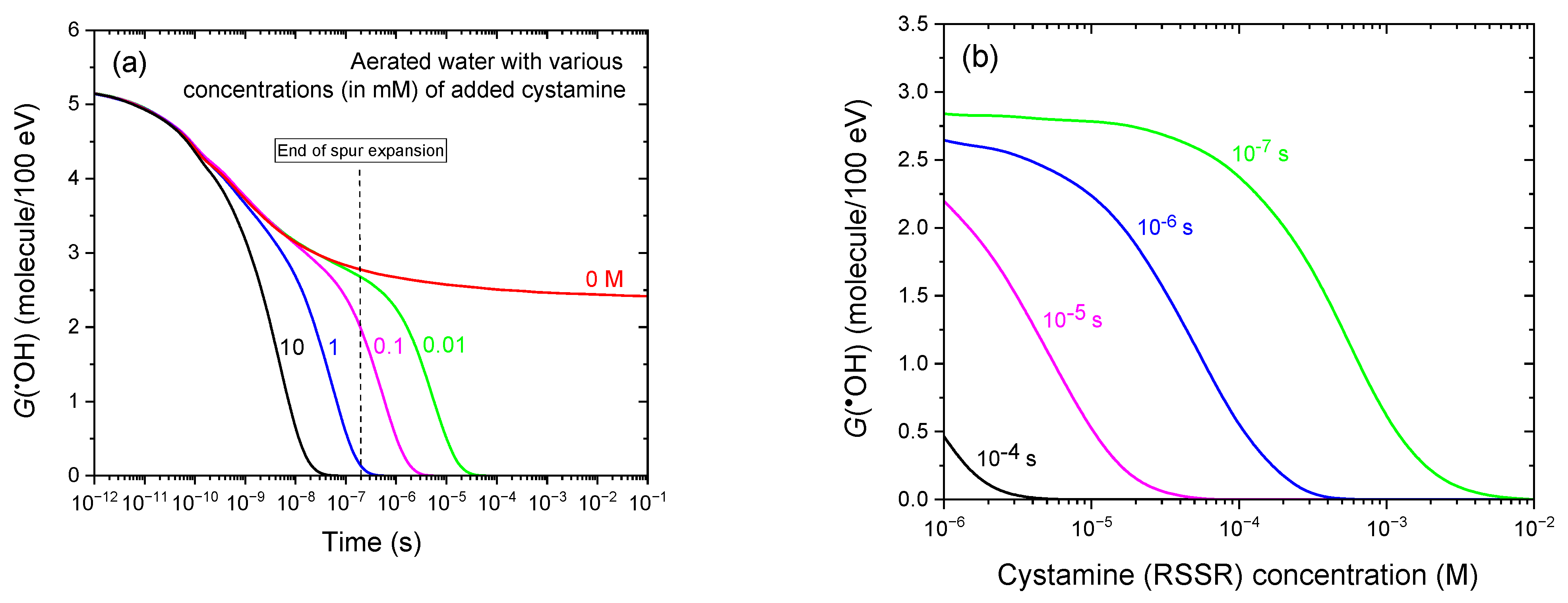
图4 (a)基于IONLYS-IRT蒙特卡罗径迹化学模拟的25℃下300 MeV质子(LET≈0.3 keV/μm)辐射有氧水时,不同胱胺(RSSR)浓度(单位:mM)下•OH产额随时间(1 ps-100 ms)的演变:10(黑色曲线)、1(蓝色)、0.1(品红色)、0.01(绿色)。红色曲线代表未添加胱胺时的G(•OH)。作为参考,约0.2 μs处的垂直虚线表示溶液从非均匀射束动力学向本体均相动力学的转变[53]。(b)为图4a的另一种呈现形式,展示了蒙特卡罗模拟得出的辐射后不同时间点(10⁻⁷、10⁻⁶、10⁻⁵、10⁻⁴ s)下,G(•OH)随RSSR浓度(10⁻3-10 mM)升高的下降趋势。模拟中采用的溶解氧浓度为0.25 mM。
具体而言,该清除过程的特征时间尺度可通过准一级速率常数的倒数估算,即1/(k[H₂]),其中k=4×10⁷ M⁻1s⁻1。由此计算得出的时间常数范围为:0.01 mM H₂时约为2.5 ms,10 mM H₂时约为2.5 μs。值得注意的是,在所有研究的H₂浓度范围内,•OH的清除主要发生在辐射作用的均相化学阶段,即辐射径迹消散之后。这一观察结果表明,H₂对•OH的辐射防护作用主要在初始生成的活性物质扩散并在本体溶液中均匀分布后才得以体现。
图3b以另一种形式呈现了图3a的结果,展示了辐射后不同时间点(0.1、1、10、100 μs)下,G(•OH)随H₂浓度(10⁻3-10 mM)升高的下降趋势。蒙特卡罗模拟结果证实,H₂对•OH的清除作用具有浓度依赖性。
3.4 氢气(H₂)与胱胺(RSSR)的抗氧化及辐射防护效率对比
图4a展示了在与图3a相同的条件下(300 MeV质子辐射有氧水),添加不同浓度胱胺(0.01-10 mM)时G(•OH)随时间的演变。与图3a、b类似,图4b以另一种形式呈现了图4a的结果,展示了辐射后不同时间点(0.1、1、10、100 μs)下,G(•OH)随胱胺浓度(10⁻3-10 mM)升高的下降趋势。蒙特卡罗模拟结果表明,胱胺对•OH具有强效且浓度依赖性的清除效率。
与H₂的作用类似,添加胱胺会因•OH与胱胺的清除反应(反应(13))[42,75,76],导致•OH产额显著且逐步下降。随着胱胺浓度的升高,•OH产额的下降趋势愈发陡峭,且起始时间逐渐提前:G(•OH)下降的起始点从0.01 mM胱胺时的约100 ns提前至10 mM时的约100 ps——这一现象反映了胱胺对•OH的高反应活性[24,42,48及其中引用的文献]。从辐射化学角度来看,值得注意的是,在所有研究的浓度范围内,胱胺对•OH的清除主要发生在辐射作用的非均匀化学阶段,早于H₂的作用时间(H₂主要在均相阶段发挥作用)。
对浓度均为10 mM的H₂和胱胺的抗氧化效率进行直接对比,可进一步凸显二者的差异:在胱胺存在时,G(•OH)从约100 ps时的4.4 molecules/100 eV急剧下降,至约30 ns时几乎降至零;而在H₂存在时,•OH产额的下降起始时间更晚(约30 ns时为2.8 molecules/100 eV),且直至约15 μs时才降至零。这些结果明确表明,胱胺清除•OH的效率显著高于H₂:其反应速度更快,能在更早的时间点清除•OH,即使在较低浓度下也是如此——这凸显了胱胺更优异的抗氧化效率和辐射防护潜力。
尽管H₂的清除效率略低于胱胺,但它具有一项独特优势:无毒性。胱胺虽然能高效清除•OH,但其潜在的细胞毒性限制了其广泛应用——尤其是在生物医学和治疗领域,安全性至关重要。相比之下,H₂兼具可观的抗氧化性能和优异的安全性,在发挥清除作用的过程中不会对细胞结构或功能造成损害[45]。
4. 讨论、结论与展望
我们的蒙特卡罗径迹化学模拟定量表明,在模拟细胞环境的条件下,低线性能量转移(LET)辐射后,氢分子(H₂)可显著降低羟基自由基(•OH)的产额,且不会明显改变其他辐射分解产物的产额。随着H₂浓度的升高,其对•OH的选择性清除速度逐渐加快。值得注意的是,即使在H₂浓度低至0.01 mM(远低于实验或临床研究中常用的0.3-0.78 mM范围)时,H₂仍能有效清除几乎所有的•OH。然而,与含硫基团介导的自由基清除能力已得到充分证实的胱胺相比,H₂中和活性氧(ROS)的效率更低。胱胺的清除效率显著更高,能在更早的时间点、以更低的浓度实现•OH的完全清除。从辐射化学角度来看,胱胺的高反应活性使其具有更优异的辐射防护性能。
但这种更强的化学效能伴随着重要的权衡:已知胱胺在高浓度下会产生细胞毒性,这限制了其在临床应用中的适用性。相比之下,H₂的核心优势在于生物安全性——这与实验结果一致。H₂无毒性,在生理条件下化学性质稳定,且能轻松穿透细胞膜。这些特性使H₂在发挥抗氧化作用的同时,不会干扰正常的细胞功能,因此在敏感场景的医疗应用中极具吸引力,例如在放射治疗中保护健康组织,或治疗与氧化应激相关的疾病。
除直接清除自由基外,H₂的辐射防护作用还可能涉及其他机制,例如调节氧化应激信号通路、发挥抗炎效应以及刺激细胞修复过程[77及其中引用的文献]。这些额外的生物学效应进一步增强了其作为治疗性抗氧化剂的潜力。综上,选择性、有效性与无毒性的独特结合,使H₂成为基于抗氧化剂的干预措施(包括癌症放射治疗中健康组织的保护,以及多种病理条件下氧化应激的缓解)的极具前景的候选物质。
重要的是,我们的模拟还提出了一个需要注意的问题:尽管H₂能高效清除•OH,但同时也会生成氢原子(H•)——尽管H•的反应活性低于•OH,但并非生物学惰性物质。在缺氧肿瘤组织等氧气匮乏的环境中,用于清除H•的氧化途径受限或失活,这些自由基可能会积累并参与其他化学反应,例如生物分子的还原或与过渡金属离子的相互作用,进而可能影响细胞氧化还原平衡或治疗效果。因此,H₂的放射生物学效应(尤其是在缺氧条件下)可能比以往预期的更为复杂。值得注意的是,在评估富氢水在放射生物学和放射治疗中的应用时,应谨慎考虑•OH清除与H•生成之间的平衡。
综上,作为一种选择性抗氧化剂和辐射防护剂,氢分子在有效性与安全性之间取得了良好平衡。尽管其化学效能可能不及胱胺,但其优异的生物相容性以及对毒性最强的活性氧(ROS)的选择性清除能力,使其成为基础放射生物学和临床放射生物学领域值得进一步研究的极具吸引力的候选物质。
关于H₂存在下无氧水或有氧水的辐射分解,目前我们尚未找到可作为直接基准的实验数据——即辐射后不同时间点各辐射分解产物的产额数据。此类数据的获取将具有重要价值,因为它们是在这些特定条件下充分验证模拟结果的关键。
最后,未来研究可关注两个极具前景的方向:
第一,可将模型扩展至高线性能量转移(LET)辐射研究——通过将质子能量从300 MeV降低至150千电子伏特(keV),覆盖约0.3至72.2 keV/μm的LET范围[48]。高线性能量转移(高LET)辐射预计会增加径迹内局部•OH的浓度,从而促进径迹内重组反应,重新生成水或产生过氧化氢(H₂O₂)。因此,由于(•OH + H•)和(•OH + •OH)等竞争反应的存在,H₂对•OH的清除效率可能会下降。
第二,高剂量率(尤其是放射治疗中“FLASH效应”相关的高剂量率)的影响也值得研究。与高LET辐射类似,高剂量率会提高本体溶液中•OH的浓度,促进径迹间重组反应生成水和H₂O₂,这同样可能降低H₂对•OH的净清除效率。
目前,我们实验室正在积极开展这两方面的研究,有望为辐射质量(LET)、剂量递送方式与氢分子的抗氧化及辐射防护作用之间的相互作用提供新的见解。
https://blog.sciencenet.cn/blog-41174-1499818.html
上一篇:抑郁焦虑的共同神经环路
下一篇:氧气在人体内运输的理论 之一 (氧气和血红蛋白结合)