博文
氧化应激氧化平衡和肾脏疾病,氢气的潜在作用机制
||
氧化应激氧化平衡和肾脏疾病,氢气的潜在作用机制
亮点:氧化应激(OS)在CKD的发生发展中起着至关重要的作用。 氢气选择性地捕获有害的ROS,如OH和ONOO-,同时保留有益的ROS。 氢气通过促进抗氧化酶的表达和降低OS来影响多种细胞通路。 临床前研究显示,氢气可降低OS,改善肾功能,但还需要更多的试验。
摘要:氧化应激在慢性肾病(CKD)的发展和进展中扮演着关键角色,它导致肾脏细胞损伤、炎症和纤维化。然而,目前缺乏有效的治疗干预措施来减缓CKD的进展。分子氢(H2)的多面药理作用使其成为一个有前景的治疗途径。氢气能够捕获有害的•OH和ONOO-,同时保持对细胞信号传导至关重要的反应性氧种(ROS)。管理细胞氧化还原平衡的NRF2-KEAP1系统可用于治疗CKD。氢气激活这一路径,加强抗氧化防御并清除ROS以对抗氧化应激。氢气可通过使用Wnt/β-catenin通路改善NRF2信号传导,并在线粒体中间接激活NRF2-KEAP1。此外,氢气通过调节细胞氧化还原状态、抑制MAPK通路和维持Trx水平来调节NF-κB活性。用氢气治疗还通过中和ROS而减弱HIF信号传导,同时间接增强HIF-1α功能。而且,氢气影响FOXO因子并增强抗氧化酶的活性。尽管台架研究的结果令人鼓舞,但临床试验仍然有限,需要进一步调查。本综述的重点是氢气在治疗肾脏疾病中的作用,特别关注氧化应激和氧化还原信号调控,以及其潜在的临床应用。
Zheng CM, Hou YC, Liao MT, Tsai KW, Hu WC, Yeh CC, Lu KC. Potential role of molecular hydrogen therapy on oxidative stress and redox signaling in chronic kidney disease. Biomed Pharmacother. 2024 May 24;176:116802.
1.引言
慢性肾病(CKD)是一种以肾功能逐渐衰退为特征的进行性疾病。其病理生理学核心是涉及反应性氧和氮物种(ROS和RNS)的氧化还原信号传导,这些物种调节细胞功能[1]。在CKD中,失调的氧化还原信号传导由于过量的ROS/RNS和抗氧化剂不足而导致氧化应激,进而引起细胞损伤、炎症和纤维化,使肾功能恶化。这种失衡导致脂质过氧化、DNA损伤和蛋白质修饰,触发损害肾功能的炎症和纤维化反应。此外,氧化应激还影响其他途径,如内皮功能障碍、高血压和动脉粥样硬化,形成肾脏和系统性损伤的循环[2]。理解CKD中的氧化还原信号强调了抗氧化疗法恢复氧化还原平衡、减少氧化应激并可能减缓疾病进程的潜力[3]。
过多的ROS和RNS是在促氧化剂和抗氧化剂失衡时产生的,导致氧化应激(OS)。肾脏疾病中常见的抗氧化水平降低和ROS产生增加是由于肾功能受损和过度产生所致[4]。O2●-(超氧阴离子)、过氧化氢(过氧化氢)和●OH(羟基自由基)是通过不同的途径产生的,如线粒体功能障碍、NADPH氧化酶激活和炎症诱导的氧化应激[5]、[6]。长期以来,ROS被认为是影响细胞信号传导和功能的特定分子调节因子[7]。这些ROS导致肾血管内皮功能障碍,造成血管舒张障碍和收缩,最终加剧肾脏损伤[8]、[9]。此外,ROS直接损害肾细胞,导致小管间质纤维化、肾小球硬化和肾功能丧失[10]。像CKD中的尿毒症毒素,例如吲哚硫酸盐,加剧了炎症和氧化应激,恶化了肾脏损伤[11]。CKD中的持续炎症进一步增加了ROS的产生,形成了一个有害的循环[12]、[13]。氧化应激通过激活NF-κB(核因子κ轻链增强子激活的B细胞),随后触发促炎细胞因子的产生,从而进一步加剧CKD的炎症[14]。ROS通过激活TGF-β(转化生长因子β)信号通路、刺激肌成纤维细胞活化和增加细胞外基质沉积等方式促进肾纤维化,导致进行性肾疤痕和功能下降[15]、[16]。此外,氧化应激破坏了自噬,这对于维持肾脏稳态至关重要,进一步恶化了肾脏损伤并加速了CKD进展[17]。
研究表明,大多数电离辐射损伤源于水的分解产生的羟基自由基(•OH)。氢气通过清除•OH和增强基因表达提供放射保护[18]。通过类似的机制,氢气疗法在缓解阿尔茨海默病病理方面也显示出希望[19]。对于慢性肾病存在多种治疗方式,包括药物治疗、肾脏透析和移植。抗氧化疗法通过清除ROS、恢复氧化还原平衡和保护肾功能,在CKD管理中具有潜力[20]、[21]、[22]。最近,利用氢气治疗这些病症的兴趣日益增加。氢气展现出强大的还原能力,通常用作生化反应中的还原剂。本综述的重点在于检查CKD中的氧化应激和氧化还原信号传导,同时探索氢气作为治疗方法的潜力。
2.氢分子在临床实践中的观点
直到2007年,人们一直认为氢气在生理上不活跃,直到发现它通过清除●OH和ONOO−来保护大脑免受缺血再灌注(I/R)损伤[23]。临床试验中最常采用的氢气干预策略包括吸入氢气或氢氧混合气、饮用富氢水(HRW)、注射富氢盐水(HRS)和口服富氢珊瑚钙[24]、[25]。这些研究几乎在200种不同的人类和动物疾病模型中展示了潜在的治疗效果[26]。氢气在清除羟基自由基方面的抗氧化作用略有了解,但其确切的目标和机制仍不清楚。除了其抗氧化作用外,氢气还具有抗炎和保护线粒体功能等多种效果[27]、[28]。氢气的自由基清除特性不能完全解释其广泛的生物学效应。
在探索氢气行动潜力机制的过程中,线粒体受到了越来越多的关注。线粒体不仅负责产生ATP,还参与细胞细胞的其他重要功能[29]、[30]。CKD涉及线粒体功能障碍和氧化应激[31]、[32]。由于氢气体积小、质量轻、电荷中性、非极性和高扩散率,使其能够在不到一分钟的时间内迅速穿透细胞膜并到达亚细胞室[33]。观察到线粒体复合物I既促进了氢气(H2)的利用也促进了其产生。这一证据表明线粒体与涉及氢气的代谢过程之间可能存在关联[34]。此外,线粒体含有大量含铁卟啉的蛋白质,如细胞色素。实验发现,铁卟啉可能是氢气诱导效应的直接接受者[35]。氢气的影响扩展到细胞器如内质网(ER),研究表明它显著抑制了ER应激[36]、[37]。
2.1 细胞信号传导的氧化还原调控
氧化还原调控是指通过调节氧化还原敏感分子,特别是半胱氨酸和甲硫氨酸残基,来控制细胞信号通路的过程[38]。它在调节细胞增殖和分化中扮演着关键角色[39]。细胞信号传导通过氧化还原调控被精细调整,许多细胞内途径对细胞氧化还原平衡的变化做出反应。NOX(NADPH氧化酶)酶的激活受到激素、生长因子或细胞因子的刺激,并导致过氧化氢的产生,随后当膜相关受体被激活时,各种细胞内靶蛋白被氧化[7]。已经观察到过氧化氢能修饰受体本身的特定半胱氨酸残基,如胰岛素受体(IR)和表皮生长因子受体(EGFR)及其在相关途径中的下游目标[40]、[41]。通常,氧化会阻碍磷酸酶的功能,而根据特定的激酶、氧化位点和活性氧物种(ROS)的水平,它要么增强要么减弱激酶的活性[40]、[42]。PTP1B(蛋白质酪氨酸磷酸酶1B)的半胱氨酸氧化使其失活,这对于维持胰岛素信号传导的有效性至关重要[43]。细胞氧化还原状态的变化直接控制AKT(蛋白激酶C)并间接影响其下游目标,包括像GSK3β(糖原合成酶激酶-3β)和IKK(IkappaB激酶)这样的酶,它们调节转录因子的活性[44]。
氧化还原调控影响基因表达、逆向信号传导、应激反应和蛋白质功能。硫氧还蛋白(Trxs)和谷氧还蛋白(Grxs)维持细胞氧化还原平衡[45]。AMPK(AMP激活蛋白激酶)调节细胞能量代谢,当低营养或高能量需求时ADP(腺苷二磷酸)/ATP(腺苷三磷酸)比率和AMP(腺苷一磷酸)水平上升时被激活。活性氧物种通过半胱氨酸残基的S-谷胱甘肽化激活AMPK[46]。此外,证据表明AMPK活性可以通过二次过程间接受到氧化还原调控的影响[47]。除了胰岛素信号传导外,各种重要的细胞内途径、酶和转录因子(TFs),如TOR(雷帕霉素靶蛋白)、JNK(Jun-N末端激酶)和STATs(信号转导子和转录激活子),都受到氧化还原调控[40]、[48]、[49]、[50]、[51]、[52]。
2.2 细胞内ROS的来源
线粒体是细胞ROS的重要来源,在应激和病理情况下产生增加[53]。早期研究表明,在最佳条件下分离的线粒体表现出高ROS产生,但这可能不反映完整细胞中的生理水平[54]、[55]、[56]。线粒体富含抗氧化剂,贡献了相当一部分细胞ROS,约占细胞抗氧化剂的三分之一[57]。用抗氧化剂靶向线粒体有效对抗与氧化应激相关的疾病,确认了它们在ROS产生中的作用[58]。线粒体抗氧化剂减少氧化应激,尽管其他细胞途径也可能有贡献[59]、[60]。增加的ROS产生依赖于NAD(烟酰胺腺嘌呤二核苷酸)和泛醌的氧化还原电位,不仅仅取决于线粒体呼吸或ATP需求的增加[61]、[62]。线粒体的ROS产生随ATP需求变化而变化,增加的呼吸减少了ROS的产生[63]。
各种细胞部位,包括细胞器,产生活性氧物种(ROS)。过氧化物酶体由Christian de Duve于1965年发现,通过产生过氧化氢的氧化酶和过氧化氢酶产生ROS,占细胞耗氧量的多达20%[64]、[65]。大约45%的细胞活性氧物种(ROS)产生归因于微体,即内质网的部分[66]。NOX家族异构体主要在细胞膜外部产生活性氧物种(ROS),也存在于各种细胞内膜中,如线粒体[67]。此外,像黄嘌呤氧化酶、细胞色素P450单加氧酶、髓过氧化物酶和环氧合酶等酶也有助于ROS的产生[68]、[69]、[70]。
研究探索了不同生理和进化背景下的ROS产生。受损的线粒体功能可以升高ROS水平,可能在神经退行性疾病和肝纤维化等疾病中使线粒体成为主要来源[71]。然而,疾病并不总是直接影响线粒体的ROS。研究确定NOX酶是再氧化过程中主要的ROS来源,这挑战了线粒体ROS仅作为ATP生产副产品的观点[67]、[72]。了解不同细胞区室中ROS调控的重要性,强调了线粒体的显著但非主要作用,以及进一步研究的必要性[53]。
2.3 肾脏细胞器ROS的来源
尽管相对于总体重的比例很小,肾脏的氧气消耗率很高,占全身氧气消耗的7%[73]。此外,其线粒体密度仅次于心肌[74]。这凸显了理解肾脏与ROS关联的重要性。在肾脏中,线粒体和NOX家族对于产生内源性活性氧物种(ROS)至关重要。
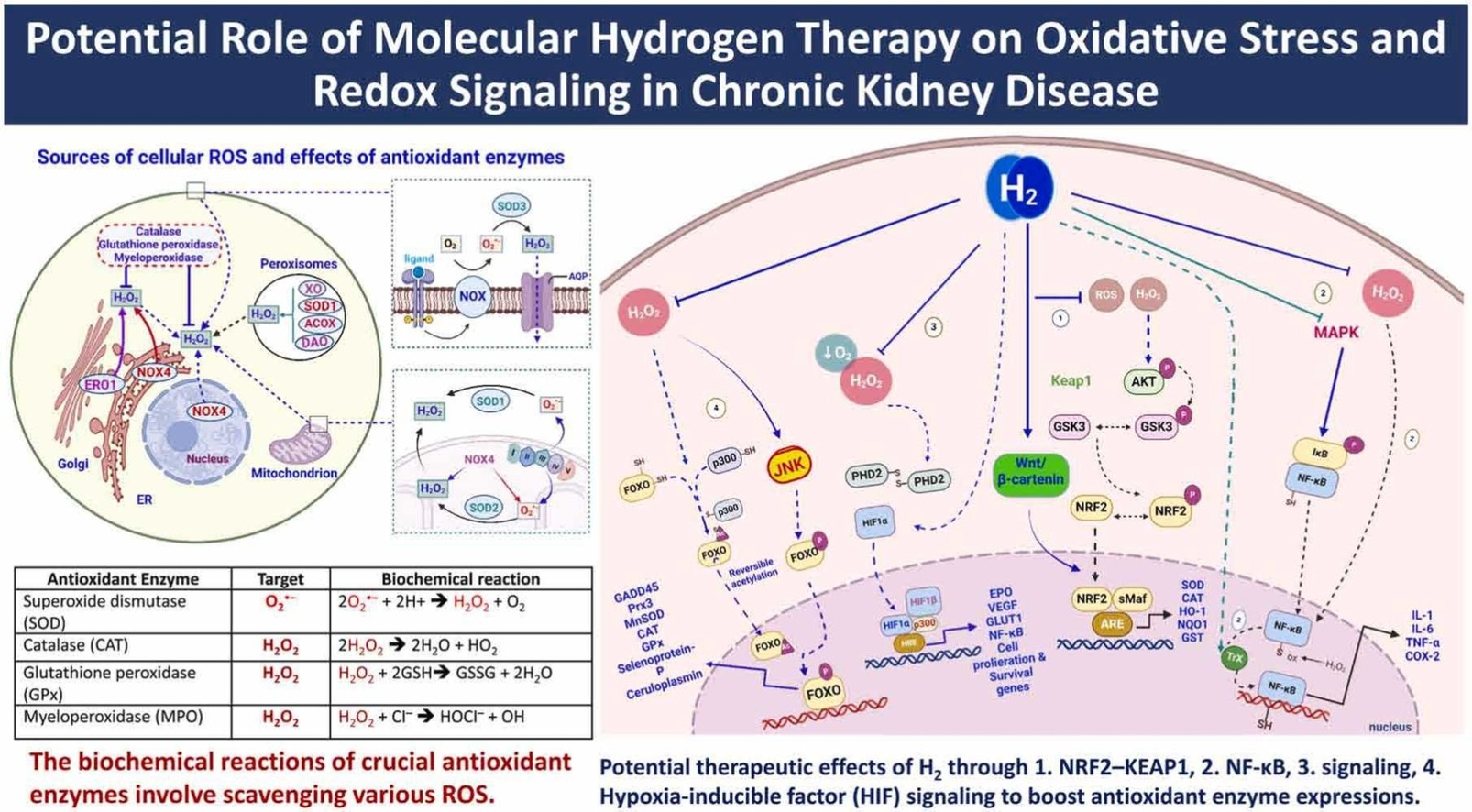
图1. 细胞ROS的来源和抗氧化酶的作用
线粒体作为关键的氧气消费者,通过多种机制产生过氧化氢。此外,像内质网(ER)、过氧化物酶体和溶酶体这样的细胞器也可以产生过氧化氢。ERO1、SOD、NOX和XO等酶和酶复合物参与了这一过程。水通道蛋白(AQP)帮助过氧化氢穿越膜,它也可以通过脂质膜扩散。CAT、GPx和MPO等抗氧化酶防止过氧化氢积累。SOD将O2•−转化为过氧化氢和氧气。在左上角,展示了抗氧化酶如何通过生化反应清除各种氧自由基。
图2. 氢气对NRF2-KEAP1信号通路的影响。
2.4 线粒体
线粒体通过其五个内膜复合物(I-V)中的氧化还原反应来产生ATP。在复合物I中,NADH被氧化,而在复合物II中,琥珀酸发生氧化。这一过程将辅酶Q还原为辅酶Q醇。在复合物III中,辅酶Q醇被氧化以还原细胞色素c,促进电子传递给氧以产生水。这一过程建立了H+梯度,驱动ATP合成[75]。泄漏的电子还原氧气,生成超氧化物。SOD1(超氧化物歧化酶1)和SOD2(超氧化物歧化酶2)分别将超氧化物转化为氧气和过氧化氢,其中SOD1作用于膜间空间,而SOD2作用于线粒体基质。抗氧化酶GPX(谷胱甘肽过氧化物酶)和PRX(过氧化还原酶)进一步将过氧化氢还原为水,维持氧化还原平衡。过量的ROS,常见于衰老或疾病中,会导致氧化应激,氢气疗法可能减轻这种情况[75]。

图3. 氢气对NF-κB信号传导的影响。
肾脏依赖大量的线粒体来排除废物和维持流体电解质平衡,为这些关键任务提供能量。线粒体通过PGC1α(过氧化物酶体增殖激活受体γ共激活因子1-α)介导的信号适应代谢变化,并通过平衡动态和能量学来维持稳态。线粒体功能障碍与CKD的进展有关,在动物模型和患者中都可见结构和功能性改变。通过遗传或药物干预增强线粒体功能,在动物研究中减轻了肾脏功能障碍[76][77]。实验数据表明,CKD中肾脏的线粒体抗氧化防御系统受损[4][78]。众多研究表明,在肾病理进展期间,肾小球细胞中的一氧化氮(NO)可用性降低,补充NO水平对肾小球功能有益[79]。补充MitoQ(米托醌),增强线粒体的抗氧化防御,有效改善线粒体功能并减轻肾脏损伤。这些发现表明,在CKD期间,肾脏线粒体在清除活性氧物种(ROS)方面受到损害[80][81]。糖尿病小鼠的肾脏组织表现出线粒体O2•−产生减少。然而,通常与线粒体功能障碍相关的线粒体活性氧物种(mtROS)的过度产生,会导致细胞损伤并促进CKD的发展[82]。
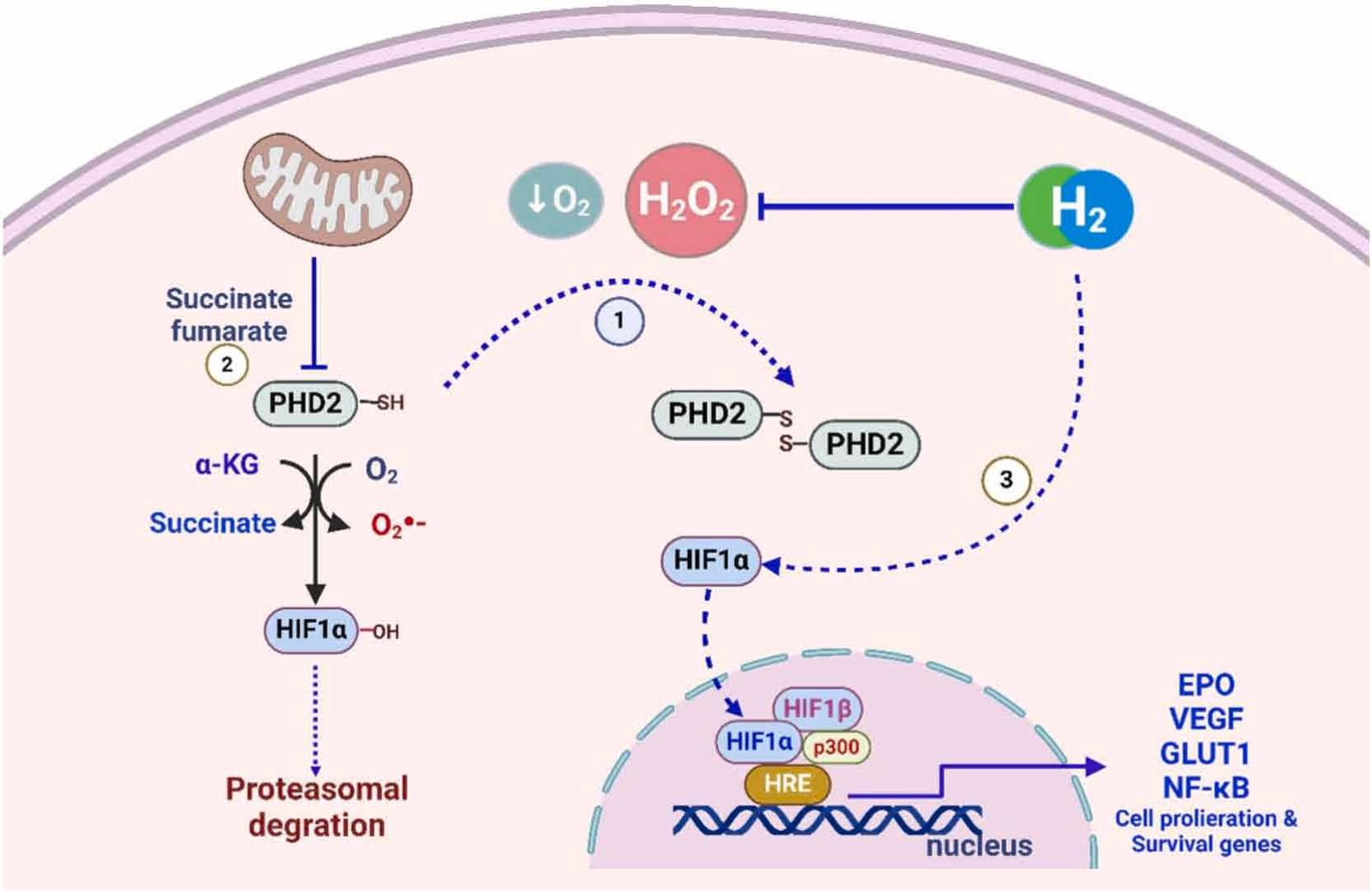
图4. 氢气对缺氧诱导因子(HIF)信号通路的影响。
2.5 内质网和过氧化物酶体
在内质网(ER)中,ERO1(内质网氧化还原酶1)主要产生过氧化氢,同时在蛋白质折叠过程中促进二硫键的形成[83]。此外,NOX4也对ER产生的过氧化氢有所贡献[84]。过氧化物酶体与线粒体和ER类似,包含CAT、SOD1和PRX5等酶,调节细胞的氧化平衡[84]。过氧化物酶体内过氧化氢的主要来源主要是通过COX介导的β-氧化作用分解脂肪酸[85]。各种过氧化物酶体酶如XO,进一步产生过氧化物酶体ROS。过氧化物酶体在肾近曲小管中丰富,而在肾小球、远曲小管和集合管中较少[86][87]。

Fig. 5. FOXOS in the cellular antioxidant defense.
2.6 产生ROS的主要酶
2.6.1 NADPH氧化酶(NOX)
NOX有七种同源物,即NOX1-NOX5、DUOX1和DUOX2,其中NOX2和NOX4在肾脏中表现出相当的表达水平[73]。NOX2,也称为NADPH氧化酶2,通过产生超氧自由基(O2•−)在先天免疫反应中发挥关键作用[88]。这种酶复合体主要存在于吞噬细胞中,包括中性粒细胞、单核细胞和巨噬细胞。激活时,NOX2产生ROS,有助于微生物杀灭和防御病原体。此外,NOX2衍生的ROS参与免疫调节,介导对自身免疫的保护和维护自身耐受性[89]。而且,NOX2可以通过调节ROS的产生来消除炎症并限制组织损伤[90][91]。在肾脏中,NOX2在管状功能中发挥作用,包括调节电解质运输和管理葡萄糖[73]。NOX2和NOX4都有助于在肾动脉内产生过氧化氢[92]。值得注意的是,在各种糖尿病小鼠模型中观察到NOX2表达升高[92][93]。使用坎地沙坦和吡格列酮降低糖尿病小鼠中的NOX2表达导致SOD水平升高,氧化应激减少,并且改善了肾脏纤维化[94]。同样,用人类重组血管紧张素转换酶2治疗糖尿病小鼠降低了血浆和肾皮质中的ANG II水平。它还降低了升高的NOX2水平,导致肾脏氧化应激减少、尿蛋白水平和肾纤维化降低。这些发现表明NOX2可能参与了糖尿病肾病的发展[73][93]。
与NOX2类似,NOX4的激活依赖于与跨膜蛋白p22phox的结合。然而,NOX4不仅局限于质膜;它还存在于各种细胞内膜中,如线粒体、内质网(ER)和核内[95]。增加的NOX4水平与一系列肾脏疾病相关,导致ROS生成增加和随后的线粒体损伤[3]。有趣的是,缺乏NOX4的小鼠在接受单侧输尿管梗阻(UUO)后表现出加重的间质纤维化和氧化应激[96],表明ROS水平过高或过低都可能导致不利结果。NOX4对肾脏功能的全面影响,无论是正面还是负面,都需要进一步研究。
2.6.2 黄嘌呤氧化酶(XO)
黄嘌呤氧化酶(XO)在嘌呤代谢中至关重要,促进次黄嘌呤转化为黄嘌呤,然后转化为尿酸。这个酶促过程涉及次黄嘌呤和黄嘌呤氧化为尿酸,同时产生ROS[69]。在哺乳动物中,XOR作为超氧离子、过氧化氢和一氧化氮的自然来源,在各种细胞途径中充当二级信使[69]。理解黄嘌呤氧化酶及其相关ROS的调节和功能对于理解它们在健康和疾病中的影响以及制定潜在治疗干预措施至关重要[97]。在哺乳动物中,它存在于肝脏、血管内皮细胞和特别是乳腺中。其主要作用是代谢核苷酸,包括腺嘌呤和鸟嘌呤[98]。此外,XOR(黄嘌呤氧化还原酶)广泛分布在哺乳动物组织中,作为乳脂中的抗菌成分[99]。在哺乳动物中,XOR转变为XDH(黄嘌呤脱氢酶)或XO[100]。XO附着在内皮细胞上,并将电子捐献给分子氧,产生O2•−和过氧化氢[101]。
XO的激活可以导致ROS过量产生和细胞功能障碍。
在一项涉及心力衰竭患者的临床试验中,使用XO抑制剂通过减少氧化应激改善了内皮功能[102]。抑制XO保护肾脏,防止损伤并保持CKD和DKD的功能[103]。它还调节胆固醇并减少ROS,有助于保护肾脏[104]。此外,XO抑制剂显示出心脏-肾脏保护作用,有益于心脏和肾脏健康[105]。总的来说,它们为保护肾脏和预防各种病症中的并发症提供了有希望的策略。
2.7 转录因子(TFs)的氧化还原调控及氢气的潜在治疗效果
2.7.1 NRF2-KEAP1和抗氧化反应
KEAP1(类Kelch ECH相关蛋白1)-NRF2(核因子红细胞2相关因子2)系统是负责调节氧化应激反应和维护细胞稳态的细胞途径。NRF2-KEAP1系统充当检测细胞内氧化和亲电性应激的传感器-效应器机制[106]。KEAP1作为NRF2的胞质抑制剂。在正常条件下,KEAP1通过泛素-蛋白酶体途径靶向NRF2进行降解[107]。当细胞遇到氧化应激时,KEAP1释放NRF2,允许NRF2转移到细胞核中。在细胞核中,NRF2与DNA中的抗氧化反应元素(AREs)结合,从而激活抗氧化和解毒基因的转录[106]。NRF2的激活导致多种细胞保护酶和蛋白质的合成,包括HO-1、NAD(P)H醌氧化还原酶1(NQO1)和谷胱甘肽S-转移酶(GST)。这些蛋白质帮助中和活性氧物种(ROS)和其他有害分子,促进细胞存活并减轻氧化损伤[108]。
NRF2触发NADPH以及硫氧还蛋白还原酶1(TrxR1)系统和GSH(谷胱甘肽)系统的合成,从而影响Trx1、硫氧还蛋白还原酶1(TrxR1)、糖皮质激素受体(GR)和GSH生物合成基因的表达[109]。TrxR1作为下调NRF2的关键因素[110]。沉默信息调节蛋白去乙酰化酶,特别是SIRT1和SIRT2,通过从KEAP1去除乙酰基来控制NRF2-KEAP1通路,从而抑制NRF2降解并促进NRF2驱动的抗氧化反应[111]。此外,氧化还原敏感的microRNAs调节NRF2[112]。
NRF2-KEAP1通路在各种疾病中至关重要。氧化应激显著影响卵巢癌。NQO1作为一种抗氧化酶,通过将醌转化为氢醌来保护细胞免受氧化应激。尽管用某些化合物处理的卵巢癌细胞中NQO1和其他酶活性增加,但这不足以防止细胞死亡。这表明NQO1的过度表达是对治疗的一种防御性但不足的响应[113]。在乳腺癌早期阶段激活Nrf2通路可降低ROS水平。然而,Keap1-Nrf2的过度活化有助于健康细胞和癌细胞抵御氧化应激和治疗效应。Nrf2在癌症中扮演双重角色,既可以抑制也可以促进细胞生长。确定其角色并战略性地针对它,在癌症治疗中具有前景[114]。前列腺癌在全球男性恶性肿瘤中排名第二。许多研究表明氧化应激在癌症的起始、进展和转移中的作用,通过活性氧物种(ROS)影响NRF2/KEAP1通路。此外,该通路是抗雄激素疗法、生化复发和放疗研究的重点[115]。在非酒精性脂肪肝病(NAFLD)中,NRF2在其进展的每个阶段——脂肪积累、炎症、纤维化和肝细胞癌发展中都起着关键作用。激活NRF2有助于管理脂质代谢和氧化应激,从而通过促进保护性基因的表达来缓解NAFLD[116]。先兆子痫(PE)以母体高血压和蛋白尿为特征。PE与增强的氧化应激和炎症有关,通常伴有低水平的抗氧化剂和升高的胎盘氧化应激。NRF2/KEAP1信号传导可能有助于缓解PE症状,通过对抗氧化应激和炎症[117]。
KEAP1-NRF2系统是治疗肾脏疾病的重要靶点[7][107]。利用动物模型和KEAP1-NRF2组分的基因修饰的研究一致揭示了NRF2在缓解由IRI(缺血再灌注损伤)、UUO(单侧输尿管梗阻)、肾毒素和糖尿病等条件引起的氧化应激中的关键作用。NRF2的积极参与减缓了与这些条件相关的肾脏疾病的进展[118][119]。这些发现强调了氧化应激在启动和进展肾脏疾病中的重要性,突出了它作为不同肾脏疾病模型中共有的通路的功能[120][121]。然而,在CKD的动物模型中,尽管肾脏存在氧化应激和炎症,NRF2活性却降低了[122][123]。NRF2活性的降低影响其目标基因,包括抗氧化酶、关键的谷胱甘肽合成酶、NADPH、HO1和主要的解毒酶NQO1[107][122][123]。此外,NRF2的激活直接与通过其对胰岛素/AKT信号响应的合成代谢过程和营养可用性相关联[124][125]。在大鼠自发发展的局灶性肾小球硬化症中也观察到NRF2活性降低的现象,导致肾衰竭[126]。这些发现共同表明,NRF2失活与启动和进展各种病因的CKD有关[122]。
对NRF2在肾脏疾病中影响的调查显示,在适当时间激活时,减缓CKD进展和减少肾纤维化的结果令人鼓舞[127][128][129]。然而,NRF2对肾小球疾病进展的影响仍然不确定,因为一些研究表明没有显著影响[130][131]。此外,在充血性心力衰竭(CHF)中激活NRF2可能导致不良反应,加剧肾脏疾病,并加重蛋白尿[132]。
通过氢气激活NRF2-KEAP1信号通路对于保护细胞免受氧化应激至关重要。氢气可以通过激活NRF2诱导抗氧化酶如NQO1和HO-1的表达[28]。氢气增强ROS中和的能力有助于减轻氧化应激,从而保护细胞免受氧化损伤和相关病理条件的影响,可能涉及Wnt/β-catenin介导的NRF2信号激活[133]。此外,氢气介导的NRF2-KEAP1通路激活在缓解线粒体功能障碍方面显示出前景[134]。
KEAP1是NRF2的天然抑制剂,指导其泛素化和降解。①通常,在正常条件下,KEAP1经历分子内二硫键形成,导致其与NRF2结合。这种相互作用促使NRF2泛素化和随后的蛋白酶体降解。②然而,当面临氧化应激、亲电体或诱导NRF2的物质时,KEAP1发生修饰,允许NRF2积累。释放后,NRF2移动到细胞核中并与sMAF转录因子结合,启动基因表达过程[118][135]。NRF2-ARE通路的激活启动了对调节抗氧化剂、转运蛋白、解毒酶、谷胱甘肽和其他细胞功能至关重要的基因的转录。关键的目标抗氧化基因包括CAT、GPX1、GSH、GST(谷胱甘肽S-转移酶)和GPx[136]。糖原合成酶激酶-3(GSK3)使NRF2磷酸化,导致其从细胞核中排除并随后降解。面对氧化应激时,GSK3对NRF2的抑制被削弱。总体而言,GSK3通过蛋白酶体控制NRF2的降解,放大了KEAP1调节的影响并影响了细胞的氧化还原状态[137]。氢气清除活性氧物种(ROS),从而减少了由氧化应激诱导的NRF2-KEAP1信号激活。相反,③氢气可能通过增强的Wnt/β-catenin信号促进NRF2信号激活。
2.7.2 NF-κB信号传导 & 炎症反应
NF-κB是一种广泛分布的转录因子,控制着免疫调节和炎症反应关键过程中的基因表达[138]。在非活跃状态下,NF-κB停留在细胞质中,与抑制蛋白结合,这些抑制蛋白是I-κB家族的一部分。激活Toll样受体的配体通过磷酸化抑制蛋白来诱导其释放[1][139]。NF-κB受到精细调控,充当炎症反应的主开关。氧化还原敏感的过氧化氢在NF-κB功能中扮演双重角色。细胞质中的过氧化氢激活该途径,而NF-κB的DNA结合区的可氧化半胱氨酸调节其活性[140][141][142]。核内过氧化氢水平的增加阻碍了NF-κB与DNA的结合,从而减少了转录活性。相反,核内过氧化物酶体增殖物激活受体γ辅激活因子1(Prx1)的水平升高刺激NF-κB活性[143]。NF-κB的激活包括一系列磷酸化事件,导致抑制剂IκB的降解。氧化还原调控通过细胞质过氧化氢激活IKK激酶和对NF-κB的p50亚基的直接修饰来实现[143][144]。IKK亚基β的Cys179的S-谷胱甘肽化导致激酶活性下降,进而减少IκB的磷酸化,结果减弱了NF-κB的激活[145]。
持续的NF-κB激活与慢性肾脏病(CKD)中的慢性炎症有关[146][147]。CKD中NRF2功能的失调导致肾脏炎症。这种失调促使氢过氧化物和脂质过氧化物在肾脏组织中积累[122][148]。像氢过氧化物和脂质过氧化物这样的NF-κB强效激活剂的存在,在NRF2功能障碍与持续NF-κB激活之间建立了明显的联系,揭示了CKD炎症、氧化应激中这些信号通路之间的相互作用。
研究表明,氢气有潜力减弱NF-κB的激活,提供了一种缓解炎症的可能手段[149]。此外,它影响其他炎症信号途径,包括急性期反应和STAT3通路,同时降低上游调节基因如CD14抗原基因的表达[149]。氢气的抗氧化特性可能间接影响NF-κB信号传导,通过减轻氧化应激[28]。此外,氢气触发NRF2-KEAP1-ARE信号通路,这不仅在维持细胞氧化还原平衡中发挥关键作用,还促进对细胞应激的适应性反应。分子氢气已被观察到可以抑制作为NF-κB上游调节器的MAPK(丝裂原活化蛋白激酶)信号通路。氢气对MAPK级联反应的这种干扰可能会中断NF-κB的激活及其后的炎症反应[150]。
NF-κB在氧化应激中的作用是复杂的。ROS可以激活或抑制NF-κB,影响炎症和免疫反应[151]。在正常情况下,转录因子NF-κB与抑制剂IκB形成复合体,并停留在细胞质中。在氧化应激的情况下,①IκB激酶(IKK)的激活导致NF-κB抑制剂的磷酸化。这触发了基于多泛素化的蛋白酶体降解IκB,释放NF-κB。释放后,NF-κB迁移到细胞核,附着在DNA反应性κ元素上,并促进前炎基因产品的转录[136][152]。②为了激活,NF-κB的DNA结合域需要由核硫氧还蛋白(Trx)还原一个氧化还原敏感的硫醇。③当IKK亚基β发生S-谷胱甘肽化时,会导致抑制性κB激酶(IKK)失活,从而减少NF-κB活性[153]。NF-κB的重要靶基因包括IL-1β、IL-6、MCP-1和iNOS等。氢气清除ROS,控制细胞氧化还原平衡,这降低了NF-κB的激活和炎症基因的表达。此外,研究表明,氢气激活NRF2-KEAP1-ARE通路,同时抑制MAPK通路。这些作用进一步减少了NF-κB信号传导,同时提高了Trx水平,从而增强了NF-κB信号传导。
2.8 缺氧诱导因子(HIF)与缺氧相关的氧化应激
缺氧诱导因子(HIF)信号传导通过α和β亚基的二聚体复合物适应低氧环境[154]。在正常氧水平下,PHDs降解HIF-α。在低氧水平下,HIF-α累积,移动到细胞核,与HIF-1β结合,并通过HREs调节基因表达,影响细胞功能,包括血管生成、红细胞生成、糖酵解和细胞存活[155][156]。HIF作为低氧的主要转录反应控制器,指导细胞对低氧进行调整[157][158]。与线粒体ETC抑制相关的缺氧导致O2•−增加和随后的过氧化氢生成。慢性间歇性缺氧激活氧化还原信号,有助于系统和细胞反应[159][160]。氧化剂在缺氧期间稳定HIF中起重要作用,促进强烈的低氧反应[161][162]。即使在常氧条件下,氧化剂也影响HIF通路。它们干扰脯氨酰羟化酶,停止其功能,并增强HIF驱动的转录活性[163][164]。HIF1是主要的异构体,负责广泛的低氧反应,而HIF2以更有针对性的方式操作,主要影响特定基因转录,如促红细胞生成素(EPO)。已经确定,在缺氧的肾脏中,HIF1α主要位于肾小管上皮细胞中,而HIF2α主要在血管内皮和间质细胞中表达[165]。在缺氧的肾脏中,HIF1α通过糖酵解而非氧化磷酸化(OXPHOS)增强能量产生[166]。在正常氧水平下,脯氨酰羟化酶PHD2羟化HIF1α,触发其降解。ROS水平的增加使PHD2失活,从而稳定HIF1α[167]。HIF的激活在急性肾损伤(AKI)中显示出保护作用。研究表明,通过抑制脯氨酸羟化酶(PHD)从而稳定HIF,可以有效减少肾小管间质损伤[168][169]。在大鼠肾移植模型中,移植前给供体使用PHD抑制剂,可防止移植物损伤,从而提高移植物的存活率[170]。在缺血性肾损伤后,HIF的激活与肾小管细胞凋亡、炎症严重程度和周围毛细血管损失的减少之间存在相关性[170][171]。系统性降低HIF2α水平会加重肾小管间质损伤,而仅在内皮细胞中重新引入HIF2α则有助于从缺血再灌注损伤(IRI)引起的肾脏损伤中恢复[172]。值得注意的是,预防性使用PHD抑制剂可以改善肾小管间质损伤,但IRI发生后使用则没有效果,这表明HIF激活在AKI治疗中的时间依赖性益处[173]。在由STZ(链脲佐菌素)诱导的1型糖尿病模型中,抗氧化治疗提高了HIF1的表达,这暗示氧化应激可能会降低HIF的表达[174]。这一观点得到一项研究的支持,该研究发现,来自透析患者的干细胞中缺氧诱导的HIF靶基因(如VEGF)的激活减弱,与无CKD个体的细胞形成对比[175]。
氢气有潜力影响抗氧化HIF信号通路。其抗氧化特性可以减轻氧化应激,从而可能通过清除ROS和减少氧化损伤来影响HIF信号通路[28]。通过保持氧化还原平衡,氢气有可能间接影响HIF活性,可能增强HIF-1α的稳定性[176]。对细胞模型的研究表明,氢气干预通过抑制HIF-1α通路增强了HT22神经细胞系中的乳酸代谢[177]。炎症可以调节HIF活性,氢气通过减弱炎症反应,可能进而减轻HIF介导的细胞对缺氧的反应[178]。
在正常氧水平下,HIF-1α通过脯氨酸残基上的羟基化被脯氨酸羟化酶(PHD2)修饰,促进HIF-1α与pVHL的识别和相互作用,最终导致其被蛋白酶体降解。①PHD2具有一个受氧化还原调控的半胱氨酸,在氧化时失活,导致形成同源二聚体,使HIF1α能够发挥作用。随后,它转移到细胞核中,然后与HIF-1β (ARNT)结合到HREs上,启动其转录活性[179]。②PHD2的活性也可以被TCA(三羧酸循环)代谢产物竞争性地阻止。受此机制影响的基因包括GLUT1(葡萄糖转运蛋白1)、VEGF(血管内皮生长因子)和NF-κB[180]。氢气通过中和活性氧物种(ROS)减弱HIF信号通路,但③它间接维持HIF-1α的功能,可能保留一定水平的HIF信号通路。氢气还通过抑制HIF-1α信号通路增强乳酸的代谢。此外,氢气通过减少炎症反应减轻了HIF介导的对缺氧的细胞反应。
2.9 FOXO & 细胞抗氧化防御
FOXO(叉头框O)系统是一类转录因子家族,调节细胞过程如新陈代谢和抗氧化防御[181][182]。当发生氧化应激时,FOXO转录因子变得活跃,提高抗氧化酶如硫氧还蛋白和过氧化物酶的水平以对抗氧化损伤[183]。与其他氧化还原信号通路类似,FOXO经历半胱氨酸氧化和上游调节机制中的氧化剂介导的改变[184][185]。FOXO通常由AKT相关的磷酸化调控,使其从细胞核移动到细胞质[184]。此外,FOXO直接受氧化还原过程的调控。值得注意的是,FOXO可以与TNPO转运蛋白形成二聚体,从而增强核转运和整体活性[186]。过氧化氢水平的升高触发乙酰化酶p300与FOXO之间的相互作用,导致乙酰化并降低DNA结合能力,这一点受到硫氧还蛋白(Trx)抗氧化系统的精细调节[187]。乙酰化不仅通过改变其对目标DNA的亲和力,而且通过影响其对磷酸化的敏感性来调节Foxo1功能[188]。然而,FOXO的可逆乙酰化能力可能部分抵消了这种对FOXO信号的影响[181][189]。
在胰岛素响应性组织中,FOXO1高度表达,对调节胰岛素信号传导和确保葡萄糖稳态具有重要作用。当与胰岛素或IGF-1受体结合时,激活一系列激酶,促使FoxO磷酸化,与14-3-3蛋白结合,并最终从细胞核中移出[190]。细胞内的活性氧物种(ROS)触发JNK的激活,进而磷酸化并激活FoxO,促进其向细胞核转运[191]。研究表明,FOXO1的遗传变异可能增加糖尿病肾病(DKD)的风险[192]。
根据其功能,FOXO目标基因蛋白可以分为四组。第一组蛋白主要与防止ROS产生有关。例如,血浆铜蛋白的主要铜蛋白蓝蛋白具有铁氧化酶活性,将Fe2+还原为Fe3+,从而减少导致脂质过氧化的芬顿样反应[193]。同样,金属硫蛋白通过螯合胞内Cu(I)也阻止此类反应[194]。第二组蛋白直接干扰活性氧物种,阻止它们与其他生物分子相互作用。值得注意的例子包括SOD2和胞质SOD1以及其他抗氧化酶如过氧化氢酶、谷胱甘肽过氧化物酶(GPx)和过氧化物氧还蛋白[195][196][197][198]。激活FOXO增加硒蛋白P的产生,一种主要的血浆硒蛋白和硒转运体[199]。第三组包括如受FOXO1调节的20S蛋白酶体等酶,参与修复氧化DNA和蛋白质[200][201]。此外,FOXOs有助于表达催化氧化蛋白还原的酶,包括硫氧还蛋白还原酶和线粒体硫氧还蛋白[202]。最后,FOXO的目标是保护性蛋白,如激活Nrf-2,刺激其表达的信号通路[203]。
氢气通过减轻氧化应激引起的抑制或通过清除自由基和抑制ROS生成来影响FOXO转录活性[204]。FOXO转录因子在抗氧化防御机制中至关重要,其活性受到细胞氧化还原状态的调节[185]。研究表明,氢气分子刺激与抗氧化防御机制相关的基因表达,并间接减弱FOXO信号通路[205]。
胰岛素/IGF信号通路和ROS精细调节FOXO的稳定性、活性和亚细胞定位[206]。①由过氧化氢刺激引起的受体酪氨酸信号激活触发AKT激酶激活,导致AKT磷酸化和激活。AKT激活后,导致FOXO失活,随后通过14-3-3蛋白促进其胞质隔离或降解。②在氧化应激下,FOXO与转运蛋白(TNPO)相互作用形成分子间二硫键,促进其进入细胞核。③在另一种情况下,FOXO可以与乙酰转移酶p300形成异二聚体,可能减弱或增强FOXO活性。④细胞ROS触发JNK的激活,导致FoxO的磷酸化和激活,促进其向细胞核转运[207]。相反,FOXOs触发编码不同细胞位置抗氧化蛋白的基因转录。而且,FOXO激活还与NRF2信号通路交叉[208]。氢气清除自由基并抑制ROS生成,减少氧化应激并影响FOXO转录活性[204]。此外,氢气增强抗氧化基因的表达,间接影响FOXO信号通路[205]。
2.10 超氧化物歧化酶(SOD)
过氧化氢(过氧化氢)在超氧化物歧化酶(SOD)的操作中具有双重作用。最初,过氧化氢可以通过氧化其必需的金属辅因子来激活SOD,这对其酶活性至关重要[209]。一旦被激活,SOD开始将超氧阴离子(O2•−)转化为过氧化氢和O2·过氧化氢,作为其他抗氧化酶的底物,这有助于调节ROS水平[210]。氢气通过各种机制显著影响SOD系统。首先,氢气抑制超氧自由基的生成,这是SOD的重要底物[211]。此外,研究表明氢气可能会增强SOD的激活,可能是通过增加SOD的蛋白表达和酶功能[212][213]。通过降低超氧水平,氢气间接增强SOD活性,这对于对抗氧化应激至关重要[214]。此外,氢气作为一种抗氧化剂,中和有害的·OH,可能进一步增强SOD活性[150]。最后,氢气的抗氧化和抗炎特性有助于细胞保护,从而提高SOD系统的整体效能[215]。
2.11 过氧化氢酶(CAT)
过氧化氢酶系统存在于大多数暴露于氧气的生物中,对于将过氧化氢分解成H2O和O2,抵御氧化应激至关重要[216]。过氧化氢酶是抗氧化酶中转换率最高的,对于代谢过氧化氢和防止氧化损伤至关重要[217]。这种酶过程防止了有害的过氧化氢积累[218],其活性依赖于其含血红素的结构,这在有氧生物中无处不在[219]。在医学上,过氧化氢酶表现出治疗与增加的氧化应激相关的疾病的潜力[220]。
在糖尿病大鼠实验中,氢气显著提高了NRF2及其下游蛋白如过氧化氢酶和HO-1的表达[221]。氢气间接支持过氧化氢酶,帮助减少氧化应激。研究表明,分子氢气激活抗氧化系统,包括过氧化氢酶,促进过氧化氢分解[215]。此外,分子氢气的抗炎和心脏保护效果进一步帮助过氧化氢酶对抗氧化损伤[222]。
2.12 谷胱甘肽过氧化物酶(GPx)
GPx系统中的酶通过减少过氧化氢和有机氢过氧化物来保护细胞免受氧化伤害[223]。GPx使用过氧化氢作为底物,对于中和ROS和脂质氢过氧化物以维持细胞稳态至关重要[223][224]。氢气通过增强其抗氧化能力和促进GPx活性,对GPx功能产生积极影响[215]。这种GPx活性的增强有助于清除ROS和脂质氢过氧化物,保护细胞免受氧化损伤[225]。此外,氢气疗法可能缓解由氧化损伤引起的GPx耗竭,加强细胞对氧化应激的防御[226]。在乙醇诱导的脂肪肝小鼠进行的研究表明,富含氢的水(HRW)减少了氧化损伤并恢复了抗氧化酶的活性[227]。总的来说,氢气支持GPx系统,增强细胞抗氧化能力,保护免受氧化损伤。
2.13 髓过氧化物酶(MPO)
髓过氧化物酶(MPO),一种血红素过氧化物酶,在骨髓发育过程中形成,并存在于白细胞嗜天青颗粒中。与NOX2产生的O2●−不同,MPO生成次氯酸(HOCl-)和其他氧化剂,帮助吞噬细胞消灭病原体[228]。中性粒细胞胞外陷阱的形成,对于捕获和消除细菌至关重要,这一过程由MPO产生的氧化剂促进[229][230]。此外,MPO作为各种疾病中的活跃生物标志物,与炎症和氧化应激有关[231]。MPO-过氧化氢-氯离子系统被认为是实验性肾脏疾病发展和进展的重要病理生理贡献者,特别是影响肾小球和肾小管间质区域[232]。氢气显著影响MPO系统,通过减轻氧化应激和增强抗氧化机制。它直接减少氧化应激并诱导如HO-1等抗氧化系统[215]。中和自由基及其在生物液体中的活性调节能力,凸显了其对MPO系统的积极影响[233]。本质上,氢气增强了MPO系统对抗氧化应激和炎症的能力,从而保护肾细胞。
2.14 氢气对线粒体氧化还原调控的生物学效应
线粒体易受损害,依赖于分子和细胞器质量控制机制以维持稳态。这些控制的丧失导致线粒体功能障碍、细胞死亡和器官衰竭[234]。氢气靶向线粒体的机制涉及与•OH和ONOO−的最小直接反应,而是主要间接激活NRF2-KEAP1抗氧化系统[134]。
氢气通过调节参与线粒体质量控制的各种途径来减少线粒体损伤[235]。实验证据提出了氢气展现抗氧化效果的三个潜在途径。首先,它直接清除•OH和ONOO−,尽管在反应常数和剂量上存在挑战[23]。其次,它可能通过在线粒体的Q腔内激活来抑制线粒体ROS(mtROS)的产生,防止电子泄漏和ROS产生[236][237]。此外,它通过提高抗氧化酶水平来增强细胞抗氧化能力,可能是通过激活NRF2介导的抗氧化途径[221]。尽管有证据支持氢气能够减少mtROS[238],但需要进一步研究以完全理解其潜在机制。
2.15 将H₂应用于促成CKD发展的因素
最近的一篇综述文章[239]包括了一个表格,总结了氢气对各种肾脏疾病治疗潜力的研究。在这里,我们分析了过去十年关于CKD相关研究的关键研究,以阐明氢气疗法对CKD患者的可能影响(见表2)。
为了寻找正在进行的临床试验,请在以下在线平台上使用关键词如氢、分子氢、肾脏、急性肾损伤和慢性肾病进行搜索:ClinicalTrials.gov(由国家卫生研究院维护的全球临床试验数据库)、EudraCT(欧盟药品管理局临床试验数据库)和ICTRP(世界卫生组织国际临床试验注册平台)。未发现使用氢气作为CKD治疗方法的临床试验。H2(气体)吸入试验在之前的临床研究中被认为不方便,而富含氢的水(液体)对于CKD患者来说相对不耐受,因为所需的水量很大。最近关于口服富含氢的珊瑚钙(HRCC,固体)胶囊的开发[24][25]可能解决了氢气爆炸风险和CKD患者需要大量饮用富氢水的问题。通过HRCC胶囊进行的氢气治疗可能对CKD管理显示出希望,但需要更多的随机对照试验(RCTs)来充分了解其在减缓CKD进展中的效果。
3结论
自由基的过量产生在慢性肾病(CKD)的进展中起着关键作用。为了对抗或最小化氧化应激的发展,安全有效地减少高ROS水平的病理效应的新方法是至关重要的。研究表明,氢气在治疗与CKD中的氧化应激相关的多种疾病方面有希望。目前,主要的建议是氢气主要通过调节氧化还原信号传导、修改抗氧化基因表达起作用,并且尽管存在争议并需要进一步研究其生物学意义,但还具有选择性的自由基清除效果。
NRF2-KEAP1系统在管理氧化还原平衡中起着关键作用。激活NRF2在治疗肾脏疾病方面显示出希望。氢气启动NRF2-KEAP1途径,增强抗氧化酶的表达。氢气清除活性氧物种(ROS),抑制由氧化应激引起的NRF2-KEAP1激活。相反,它可能通过Wnt/β-catenin增加NRF2信号。氢气在线粒体中间接激活NRF2-KEAP1,减少了与ROS如•OH和ONOO−的直接相互作用。NF-κB调节免疫反应、炎症和细胞生长。氢气通过清除ROS和调整细胞氧化还原状态来调节NF-κB。氢气激活NRF2-KEAP1-ARE并抑制MAPK途径,降低NF-κB信号但增加Trx水平。HIF协调对低氧水平的反应,诱导红细胞生成和血管生成。氢气通过中和ROS同时间接支持HIF-1α功能来减少HIF信号。FOXO因子维持细胞稳态;氢气通过解除氧化应激诱导的抑制或激活来影响它们的活性。抗氧化酶如SOD、CAT、GPx和MPO清除ROS;氢气增强它们的活性。
然而,氢气在CKD中的主要目标仍然不清楚。与大多数亲水性化合物不同,这些化合物留在膜中而不进入胞质,氢气可以迅速分布到胞质和细胞器中[178]。因此,氢气可能为缓解氧化应激及其相关效应提供一种新的治疗策略。关于氢气的双重效应存在担忧:虽然它抑制ROS,抑制癌症生长[246],但它也可能促进具有增强线粒体活动的癌症的增殖[247]。因此,需要进行更多调查以全面了解氢气为CKD提供有益和安全治疗方法的确切机制。
https://blog.sciencenet.cn/blog-41174-1438528.html
上一篇:氢气补充能否增强运动员表现?系统综述和荟萃分析
下一篇:白垩纪蚂蚁的触角可能会让它们使用信息素“说话”
