博文
iMeta:德布鲁因图在微生物组研究中的应用(全文翻译,PPT,视频)
||

德布鲁因图在微生物组研究中的应用
Applications of de Bruijn graphs in microbiome research
DOI: https://doi.org/10.1002/imt2.4
发表日期:2022年3月1日
第一作者: Keith Dufault‐Thompson
通讯作者:Xiaofang Jiang(江晓芳)(xiaofang.jiang@nih.gov)
主要单位:美国国立卫生研究院国家图书馆 (Intramural Research Program, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA)
图文摘要
亮点
基于德布鲁因(de Bruijn) 图的序列组装方法一直是测序方法广泛应用的一个重要部分,尤其是在微生物组研究中
德布鲁因图可以用来有效地表示测序数据,其格式具有高度的可扩展性,并且可以扩展和修改以解决不同的研究问题
基于德布鲁因图的分析方法已经开发并运用于比较基因组学、遗传变异的鉴定以及未组装测序数据的大规模搜索
德布鲁因图数据结构将继续成为未来序列组装和分析方法的核心组成部分
作者视频解读
Bilibili:https://www.bilibili.com/video/BV1tq4y1i7Df/
Youtube:https://youtu.be/3o12ppXY04g
中文翻译、PPT、中/英文视频解读等扩展资料下载,请访问期刊官网:http://www.imeta.science/
摘要
高通量测序已经成为微生物组研究日益关键的组成部分。基于德布鲁因(de Bruijn) 图的组装高通量测序数据方法的发展,已经成为更广泛地采用测序作为生物学研究的重要组成部分。关于德布鲁因图的构建和描述方面的最新进展催生了利用德布鲁因图数据结构来辅助不同生物学分析的新方法。这些方法的应用之一是测序数据组装的替代方法,如基因靶向组装(仅从较大的宏基因组中组装基因序列)和差异组装(组装两个样品之间存在差异的序列)。此外,德布鲁因图已应用于比较基因组学。它的结构特征可用于识别序列中的变异、插入缺失(indels)和同源区域,因此可用于表示海量的基因组或宏基因组集合。研究人员甚至已经开始将基于德布鲁因图的测序数据描述应用于整个测序数据库的大规模搜索和实验发现。德布鲁因图在高通量测序数据的处理中发挥了核心作用,依赖于这些数据结构的新工具的快速发展表明,它们未来将继续在生物学研究中发挥重要作用。
引言
基因组测序技术的快速发展和改进推动了微生物组研究中的重大进展,例如增加了参考基因组的可用性,提高了高通量测序技术对整个微生物组进行测序的能力。随着这些技术进步,如何管理、处理和分析这些数据(通常以短读长序列的形式出现成为了新挑战。这些挑战已通过开发新的算法和软件得到解决。在如何处理短读长测序数据方面一些最重要的进展则来自于德布鲁因图(de Bruijn graphs,DBGs)的应用。这是一种表示序列片段(称为k-mers)之间重叠关系的网络,通常从基于一组输入序列中获得。DBG在基因组组装中得到了广泛的应用,它们构成了许多最有效的基因组和宏基因组从头(de novo) 组装算法的核心组成部分。在过去的十年里,DBG作为分析工具的组成部分也被应用于各式各样的任务,包括细菌泛基因组分析、基因组变异鉴定和组学样本比较。虽然这些方法尚未作为众多微生物组研究的一部分得到广泛采用,但它们已展现出有希望的结果。DBG已经在处理短读长测序数据方面取得成效,并将随着测序在微生物研究中日益凸显的地位而继续发挥其重大作用。
正文
德布鲁因图在基因组和宏基因组组装中的应用
短读长序列的组装
在微生物组研究中,将短读长序列组装成更大的基因组序列是使用下一代测序(NGS)技术的基础。这个问题已经通过多种方法解决,这其中包括Greedy Assemblers和Overlap-layout-consensus assemblers所采用的方法——它们通常依赖于对原始读长之间的重叠区域的识别或者依赖于基于参考基因组的组装软件的使用(即将读长映射到已经组装好的参考基因组)。这些方法被广泛用于早期的基因组组装并且在今天依然得以继续使用,但是它们也存在着局限性。Greedy和Overlap-layout-consensus都使用读长之间重叠区域的信息,这两种方法计算起来可能非常耗时,而且在组装低复杂度序列(如重复序列)和处理具有高测序深度的样本时经常会遇到问题。基于参考基因组的组装可以产生高质量的基因组组装,但是这种方法需要关联紧密的生物体基因组,这就限制了其在新生物体中的应用,并且可能在解析与参考序列模糊的读长映射方面存在问题。基因组和宏基因组的短读长组装中最重要的进步来自于DBG的应用,它克服了其他组装方法的许多限制。基于DBG的组装方法不依赖于计算读长之间的重叠,从而避免了Greedy和Overlap-layout-consensus组装中所涉及的计算密集型步骤,并且它们绕过对参考基因组的需要,只需要测序读长。基于DBG的组装可能对测序错误敏感,这会给图形带来额外的噪声,但总的来说,DBG方法的优势促进了其在组装短读长基因组和宏基因组数据方面的广泛应用。
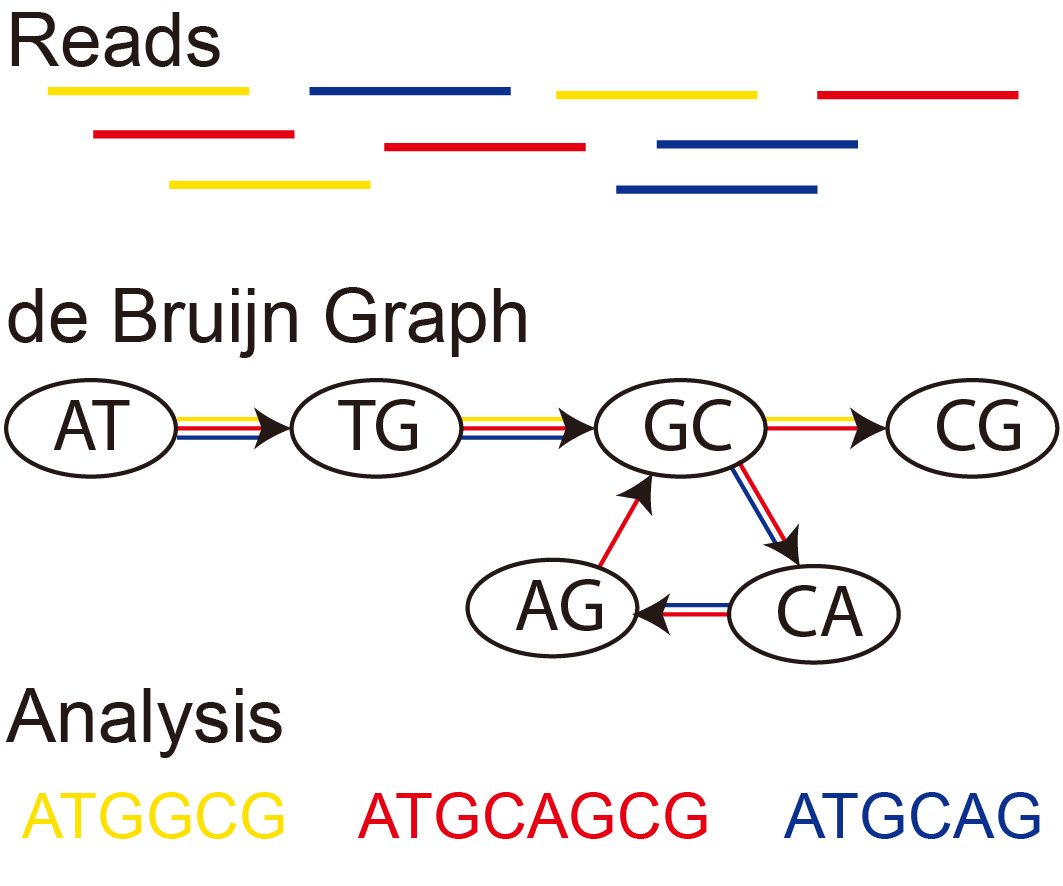
基于DBG的基因组组装起始于将原始测序读长分解成长度为k的子序列,称其为k-mers。然后通过首先为每个k-mer定义前缀,k-mer减去最后一个核苷酸和后缀,k-mer再减去第一个核苷酸来构建图。唯一后缀和前缀的总集合形成了图中的节点,并且基于链接为给定后缀和前缀的k-mers添加边。然后,通过在图中找到一个欧拉循环,即一条访问图中每条边(代表一个k-mer)一次的路径,折叠该路径中k-mer的序列来组装更长的序列,从而完成更长序列的组装(图1A)。基于DBG的基因组组装不需要计算读长之间的比对,使测序数据的组装变得有效且有可扩展性。早期的DBG组装工具,包括EULER, EULER-SR, Velvet, and ALLPATHS都采用了上述基本策略,如果对其进行修改,就可以解决特定的难题,比如,组装重复序列以及检测和处理测序错误。后来的组装方法,例如SPAdes系列软件、SOAPdenovo系列软件和MEGAHIT所采用的的方法,建立在早期组装方法的概念之上,侧重于提高效率,处理更大的数据集,如来自宏基因组的数据集,并提高组装的准确性。总体而言,这些基于DBG的组装工具代表了序列组装的重大进步,克服了许多阻碍旧组装方法的挑战,使其在微生物组研究中广泛用于序列数据的组装。
图1 德布鲁因图在基因组和宏基因组组装中的不同应用

(A) 德布鲁因图组装示意图。首先,从原始读长构建德布鲁因图,然后识别访问每个k-mer的路径(图中红色箭头),最后基于该路径组装序列;
(B) 基因靶向组装的一般过程示意图。首先使用参考序列或配置文件来识别可能包含部分基因序列的读长,然后使用该信息向图中添加权重(较粗的黑色箭头),最后使用这些加权路径直接组装基因序列;
(C) 差异组装的概念示意图。德布鲁因图由多个宏基因组生成(红色和蓝色图)。可以组合这些德布鲁因图,从而揭示两个宏基因组之间共享图的部分(灰色节点和边)或一个宏基因组独有的部分(红色或蓝色节点和边)。也可以用于组装一个样品相对于另一个样品中唯一存在的序列。
基因靶向组装
在许多微生物组研究中,期望的结果之一是鉴定感兴趣的基因,这些基因可用作系统发育的标记、疾病信号或者代表独特功能。尽管宏基因组组装方法已经显著改善但一些挑战仍然存在,包括组装过程中对微生物组群落中的优势成员的偏向性从而导致更稀有的基因被遗漏,并且宏基因组组装可能具有更高的计算成本。基因靶向组装方法则试图来解决这些问题,其策略是通过直接从宏基因组数据中组装基因序列,而不是从组装后的重叠群中预测基因序列。许多基因靶向的宏基因组组装方法在组装过程中利用德布鲁因图。这些方法通常基于隐马尔可夫模型(profile HMM)来搜索,以识别可能包含部分基因编码序列的读长。一些方法,如Xander和MegaGTA使用的方法,使用这种搜索信息向图中的特定路径添加权重来修改德布鲁因的组装图(图1B),以帮助识别和装配基因序列。其他的工具,包括SAT-Assembler,MEGAN-Assembler和phyloFlash,使用搜索结果过滤原始读长,以便在组装过程中只使用可能用于编码序列的读长。其中一些基因靶向组装方法利用了DBG的扩展版本,突出了DBG在不同类型分析中的灵活性(表1)。这些经过修改的DBG方法,包括Xander和MegaGTA使用的加权DBG图、MetaPA中使用的基于氨基酸序列的DBG图,以及MegaGTA中使用的称为简洁DBG (succinct DBG,sDBG)的DBG图变式。sDBGs是DBG的内存效率的变式,旨在应用于大型数据集,如由宏基因组和细菌泛基因组(pangenome)生成的数据集,并且已经被多种基于德布鲁因图的装配和分析方法采用,包括MegaGTA、MetaGraph和MEGAHIT。基因靶向组装可以促进宏基因组数据的分析,同时避免一些与组装过程相关的潜在偏差。这个方法不仅可以识别群落中较稀有物种的基因,还可以基于宏基因组测序提供群落中存在的生物体和基因更完整的图景。
表1 应用于基本德布鲁因图数据结构的常见改良版本以及使用它们的应用示例

从宏基因组中鉴定微生物种类
微生物组研究的共同目标之一是确定存在哪些细菌以及它们具有哪些基因。可以使用宏基因组学获得这些信息,但这需要能够区分哪些读长和重叠群来自不同物种,以便更好地理解生物体的潜在作用。Wang等人证明了德布鲁因图在鉴定宏基因组中的不同微生物菌株中的效用,其中他们将读长映射到宏基因组组装的德布鲁因图上从而区分来自不同细菌菌株的读长,而不使用参考基因组。最近的许多研究工作集中在从宏基因组读长中获得几乎完整的微生物基因组。这些由宏基因组组装的基因组(MAGs)是由分箱组装的重叠群基于核苷酸频率和读长覆盖率生成的,其依据是基于这些因素在原始群落中的物种之间会有所不同的假设。最近研究人员在改善宏基因组分箱技术时已经尝试引入德布鲁因图以帮助组装和完善MAGs。这些方法(包括GraphBin和METAMVGL)整合了 DBG的结构特征,如k-mers之间的连接和图中不相连的部分,从而细化每个MAG中包含的重叠群。这些方法突出了DBG在下游分析中的效用。在下游分析中,DBG已经存在的信息可用于改进后续分析,并可极大地提高更高质量MAGs的组装。
组学样本的比较和差异组装
随着宏基因组测序变得越来越便宜,它正在成为一种更常用的方法,并且研究通常会涉及对多个宏基因组进行测序。这就需要有效的方法来识别来自不同样本的宏基因组之间的相似性和差异性。尽管如此,这些数据的规模和复杂性使其成为一项艰巨的挑战。最近的研究提出了基于DBG的方法来进行这些比较。EMDeBruijn利用从多个微生物组数据中生成的德布鲁因图,并应用统计学方法来比较不同样本之间的距离。这种方法已被用于观察病毒群体并帮助确定丙型肝炎传播的特征,证明了其在不同类型的生物学分析中的实用性。同样地,MetaFast使用由多个宏基因组构建的简化DBG来量化相似性,提供了一种比较不同环境或样本之间多样性的方法。最近提出的MetaGraph方法显示出巨大的前景,允许以基于高效DBG的格式对整个测序数据库或多个宏基因组进行索引和查询。这种方法的一个广泛应用就是作者所说的“差异组装”。在这种方法中,可以使用MetaGraph DBG来识别在某些宏基因组中发现的k-mers,但不能识别其他宏基因组中发现的k-mers。之后可以对这些宏基因组进行组装和分析,以观察样本之间微生物群落的差异(图1C)。这些用于比较宏基因组的方法不需要在样本之间进行额外的读长映射或使用参考数据集,因而具有广泛的应用,并使组学样本间的比较更为高效和准确。
使用德布鲁因图的比较基因组学和宏基因组学
使用有色德布鲁因图的比较基因组学
微生物间遗传变异的识别,如单核苷酸变异(SNVs)和插入缺失(indels),在生物医学和生态学研究中在监测病原体的爆发以及在菌株水平上区分微生物组都有着广泛的应用。用于检测遗传变异的许多标准方法使用到参考基因组或序列的映射,但是这会使得计算更为耗时,并且当参考基因组不可用或差异太大而不能用于精确比较时,方法就不适用了。为了解决这个问题,已经开发了多种使用DBG的工具来进行无参考基因组的变异检测。这些方法是一种DBG变式,通常被称为有色德布鲁因图的DBG (cDBG),其是从多个来源(例如,多个基因组或不同的宏基因组样本)构建的DBG图,并根据k-mers是否存在于某个样本而为其分配不同的“颜色”注释(图2,表1)。研究人员已经创造了多种工具来辅助从基因组集合或多组原始读长中构建这些cDBGs,包括TwoPaCo、Bifrost,Cuttlefish。构建这些DBG的能力也促进了多种其他工具的发展,这些工具致力于在不依赖参考基因组的情况下使用DBG来鉴定遗传变异体。
图2 有色德布鲁因图的概念以及使用该图进行变异识别的过程
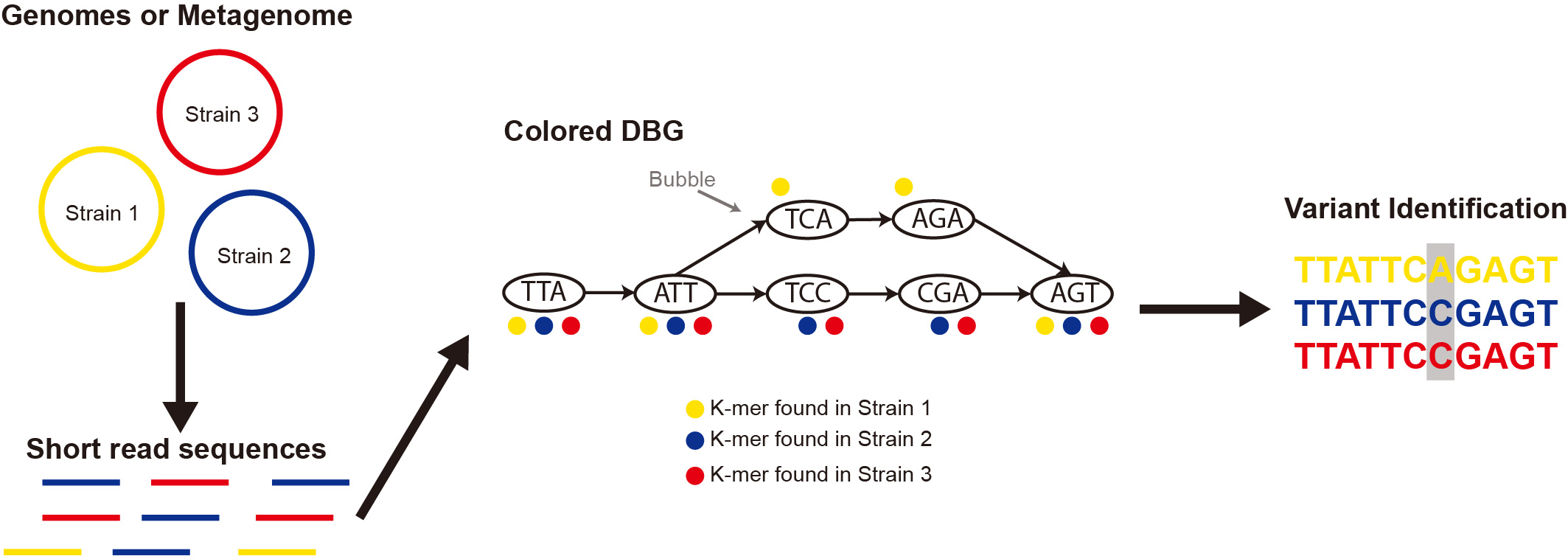
这些工具大多已开发用于鉴定遗传变异,或基于一组组装的基因组,或基于对同一物种的不同个体测序的原始读长。为了检测SNV,Cortex、MCCortex、DiscoSnp和Bubbleparse等工具都是基于对DBG或cDBGs的分析而开发的,以识别通常在图中称为“气泡(bubbles)”的结构特征。这些结构特征是由不同k-mers形成的平行路径分叉然后又汇合的点(图2),在其中就可能包含SNV。这些概念随后被扩展到工具DiscoSnp++ 和Scalpel中以检测更复杂的遗传变异,如小的插入和缺失。这一流程在BubbZ方法中得到了延生,此方法利用DBG的压缩形式来检测基因组之间的同源区域,从而允许在不需要全基因组比对的情况下进行不同基因组之间的比较分析(表1)。这些方法在微生物组研究中有许多潜在的应用,因为微生物组研究通常缺乏许多微生物菌株的高质量参考基因组,而无需参考基因组的方法将为分析微生物组和分离株开辟多条新的途径。
宏基因组中的无参单核苷酸变异鉴定
与处理基因组相比,识别宏基因组样本中的遗传变异要困难得多。宏基因组样本可能存在许多微生物物种,包含多个密切相关的菌株,不同生物体之间可能具有密切同源的基因,所有这些都使得在宏基因组中应用传统的变异鉴定方法变得困难。许多先前描述的用于变异鉴定的技术可用于或适于宏基因组数据集的分析,包括Cortex,DiscoSnp++和Scalpel基于同样的概念也可适用于宏基因组DBG。虽然这些方法可以应用于宏基因组数据,但专门用于从宏基因组中进行无参考变异鉴定的工具并不多。最近发表的LUEVARI方法利用了cDBG,其中图的着色基于宏基因组中的读长。与其他工具相比,它可以从宏基因组中更显著更灵敏地鉴定变异。随着宏基因组测序正在迅速成为研究微生物组的标准方法,这些变异鉴定方法对微生物组研究具有愈发重大的意义。
组学数据集的查询与实验发现
随着所产生的组学数据的规模和数量的增加,有效查询这些数据的方法的需求也应运而生。对已经组装的数据集执行搜索会有一些弊端:受限于搜索方法的效率,使用不同方法组装的基因组质量参差不齐,以及可用于组装的数据集较小。最近已经开发了多种方法来帮助从大数据集中构建cDBG,包括来自NCBI序列读取档案(SRA)的整个数据库,并且可用于查询这些cDBG搜索方法的后续开发使得它们可以用于大规模搜索和实验发现。这些进步的一个重要组成部分是有色DBGs紧凑版本的开发,如sDBG,Rainbowfish DBG,Cuttlefish DBG,splitMEM,还有Simpletigs DBG,它们采用各种方法来减少存储DBG和有色数据(coloring data)所需的大小、复杂性和内存(表1)。这些更有效的DBG呈现方式是高度可扩展的,这意味着它们可以有效地应用于相当大的数据集,此外研究者已经开发了多种在这些图上执行搜索的方法。Mantis和VARI程序利用基于索引的查询方法识别哪些k-mers存在于不同的序列数据集中,并且能够有效地查询SRA数据库中所有已知人类转录物的存在与否以及查询来自食品生产设施的宏基因组样品。类似地,最近提出的MetaGraph使用了k-mer匹配的搜索和基于序列到图比对的搜索方法以查询MetaGraph的索引。微生物组研究面临的主要挑战之一是实验发现,或如何在快速增长的序列数据库中找到含有感兴趣基因的测序项目。这些基于DBG的方法不仅允许将这些大型序列数据库表示为简明的cDBGs,还允许对这些索引数据集进行有效搜索,从而使得它们在微生物研究中有更广泛的应用。
德布鲁因图在转录组学和蛋白质组学中的应用
德布鲁因图也被用于分析转录组和蛋白质组数据。这些其他类型的组学数据带来了自己独特的挑战,以及用于分析它们的方法不同于用于宏基因组的方法。这些类型的组学数据的组装和分析通常依赖于参考数据库,但它们通常无法捕获代表性不足或新的转录物和蛋白质。利用DBG的方法试图通过使用配对组学数据(paired Omics data)来克服这一问题。它们使用从同一样本测序的宏基因组构建的DBG来帮助组装和分析宏转录组或宏基因组。Read2Graph依赖于将宏转录组的读长与配对的宏基因组产生的DBG进行比对,与从头开始的宏转录组组装方法相比,转录物的组装有了明显的改善。类似地,Graph2Pep和Graph2Pro方法使用配对的宏基因组或宏转录组程序,以极大地改善宏蛋白质组样本中肽的鉴定。除组装外,将读长映射到DBGs可以帮助识别剪接,以及根据RNA-Seq数据进行更准确的表达估计。宏转录组和宏蛋白质组数据的高效组装和分析一直是一个重大挑战,因此限制了以上这些方法在不同研究中的广泛应用。开发基于图的有效分析方法具有重大潜力,并且可以允许在日益复杂的生物系统中更广泛地应用多组学方法。
德布鲁因图在微生物研究中的未来作用
通过高通量测序研究微生物组已成为生物医学和环境微生物学的一个不可分割的组成部分。高效收集和分析测序数据方法的不断发展有助于在生物学研究中广泛采用测序技术,而DBG是其中许多方法的核心要素。DBG一直是短读长组装方法的重要组成部分,用于组装和分析长读长测序数据的方法已经在开发中,证明了它们在这种快速发展的技术中的应用。此外,在处理DBG的有效构建和表示方面继续取得重大的算法进展,这将为新方法的开发提供基础。虽然DBG无疑将继续在组装中发挥核心作用,但在过去十年中,它们在分析工具中的使用也在迅速增加。这些基于DBG的方法已被证明是高效且高度可扩展的,它们可以被应用于极其庞大的数据集,还可以利用不断增加的可用的组学数据开辟新的生物发现途径。随着测序成本的降低和更广泛的应用,德布鲁因图将继续成为微生物组研究中许多工具的核心。
编译:吴季秋(jwu@ucc.ie) 爱尔兰科克大学(UCC)
责编:马腾飞 南京农业大学
审核:iMeta期刊编辑部
作者简介

Keith Dufault-Thompson,美国国立卫生研究院国家图书馆博士后。Keith现阶段的研究方向为通过细菌的代谢以及生理特征研究生物体和其微生物组之间相互作用。在进入美国国立卫生研究院前,Keith于2020年在罗得岛大学取得博士学位。他研究的主题是微生物代谢网络的功能与变化。

江晓芳,美国国立卫生研究院国家图书馆首席研究员。江博士于2016年在弗吉尼亚理工大学获得遗传学、生物信息学和计算生物学博士学位,论文主题是亚洲疟疾蚊的基因组学与转录组学分析。2016年江博士进入麻省理工学院(MIT)从事博士后训练,师从Eric Alm和Ramnik Xavier。她在MIT的研究主要课题涉及可颠换元件、可遗传移动元件等在微生物组中的发现和功能识别。她于2019年加入美国国立卫生研究院国家图书馆,目前的研究工作侧重于开发和改进计算软件与算法从比较基因组学角度研究微生物组,从而为生物医学和临床健康科学中基于微生物组的诊断、治疗和预防提供支持与指导。
引文
Keith Dufault-Thompson, Xiaofang Jiang. 2022. Applications of de Bruijn graphs in microbiome research. iMeta 1: e4. https://doi.org/10.1002/imt2.4
iMeta—微生物组/生物信息高起点期刊


联系方式:
主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:office@imeta.science
微信公众号:iMeta
iMeta相关资讯
- iMeta:哈佛刘洋彧等基于物种组合预测菌群结构的深度学习方法(全文翻译,PPT,中英视频)
- iMeta:吴青龙/王明福/刘金鑫等-从肠道菌群看待人类对高原饮食的适应性(全文翻译/PPT/中英视频)
- iMeta:西农韦革宏团队焦硕等-土壤真菌驱动细菌群落的构建(全文翻译/PPT/视频解读)
- iMeta:高颜值高被引绘图网站imageGP在线发表
- 高影响力期刊iMeta扬帆起航(微生物组&生物信息)
- 如何投稿iMeta期刊?ScholarOne投审稿系统作者使用教程
- 报告视频录制:腾讯会议录屏+人像画中画特效
- Publons:文章审稿、编辑工作认证平台


https://blog.sciencenet.cn/blog-3334560-1329036.html
上一篇:MS:中山大学丁涛,吴忠道-肠道菌群调控血吸虫病传播媒介光滑双脐螺适生性的新机制
下一篇:iMeta:青岛大学苏晓泉组开发跨平台可交互的微生物组分析套件PMS(全文翻译,PPT,中英视频)