博文
R语言如何筛选数据框中的列--select
||
大家好,我是飞哥呀。
我们知道,R语言学习,80%的时间都是在清洗数据,而选择合适的数据进行分析和处理也至关重要,如何选择合适的列进行分析,你知道几种方法?
如何优雅高效的选择合适的列,让我们一起来看一下吧。
1. 数据描述
数据来源是我编写的R包learnasreml中的fm数据集。
r$> library(learnasreml) r$> data(fm) r$> head(fm)

「我们的目的:」
❝提取fm的TreeID,Rep,dj,dm,h3,并重命名为:ID, F1, y1 , y2, y3
❞
2. 使用R语言默认的方法:列选择
这一种,当然是简单粗暴的方法,想要哪一列,就把相关的列号提取出来,形成一个向量,进行操作即可。比如
r$> d1 = fm[,c(1,3,6,7,11)]
r$> head(d1)
TreeID Rep dj dm h3
1 80001 1 0.334 0.405 239
2 80002 1 0.348 0.393 242
3 80004 1 0.354 0.429 180
4 80005 1 0.335 0.408 301
5 80008 1 0.322 0.372 271
6 80026 1 0.359 0.450 258
r$> names(d1) = c("ID","F1","y1","y2","y3")
r$> head(d1)结果:

「缺点:」
这种方法,需要找到性状所在的列号,然后还要重命名,比较麻烦。
而且,后面如果想要根据列的特征进行提取时(比如以h开头的列,比如属性为数字或者因子的列等等),就不能实现了。
这就要用到tidyverse的函数了,select,rename,都是一等一的良将。
3. tidyverse的rename函数
代码:
a2 = fm %>% rename(ID=TreeID, F1 = Rep, y1 = dj, y2 = dm, y3 = h3)

这里,rename只是单独的修改名称,并没有提取出来。
还要使用select进一步的提取:

4. tidyverse的select函数
如果使用select函数,一行代码就可以搞定:
a1 = fm %>% select(ID=TreeID, F1 = Rep, y1 = dj, y2 = dm, y3 = h3)

5. select函数注意事项
「常见的坑:」
❝注意,MASS包中也有select函数,而且优先级更高,如果你载入了MASS包,select就不能使用了。
❞

哪怕你再次载入tidyverse包,也不行:

载入dplyr包,也不行:

「MASS就是这么豪横。」
像这种情况,解决办法有两种:
5.1 绝对引用函数
即使用select时,要用dplyr::select
a3 = a2 %>% dplyr::select(ID,F1,y1,y2,y3)

这样也比较麻烦。
5.2 放到环境变量中
「推荐的方法:」
r$> select = dplyr::select r$> a3 = a2 %>% select(ID,F1,y1,y2,y3)
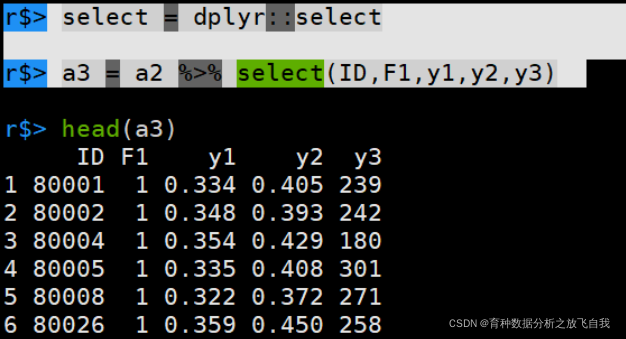
推荐在载入包时,将下面代码放在开头,就可以肆无忌惮的应用select了,毕竟,环境变量的优先级是第一位的。
library(tidyverse) select = dplyr::select
6. 提取h开头的列
这里,用starts_with,会匹配开头为h的列。
其它还有contains,匹配包含的字符,还有end_with,匹配结尾的字符。
应有尽有,无所不有。
re1 = fm %>% select(starts_with("h"))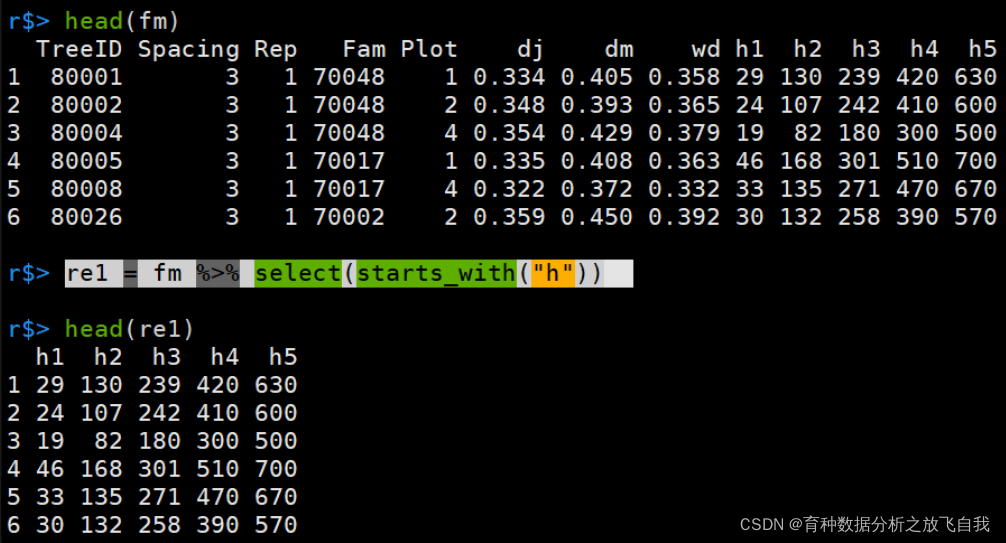
7. 提取因子和数字的列
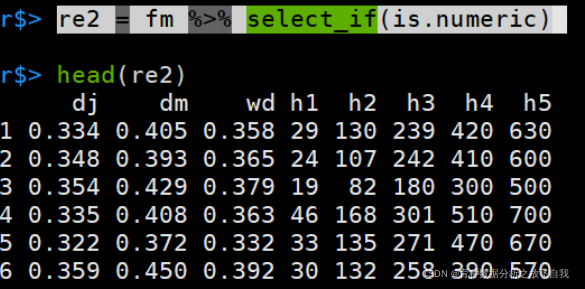
「匹配数字的列:」
re2 = fm %>% select_if(is.numeric)
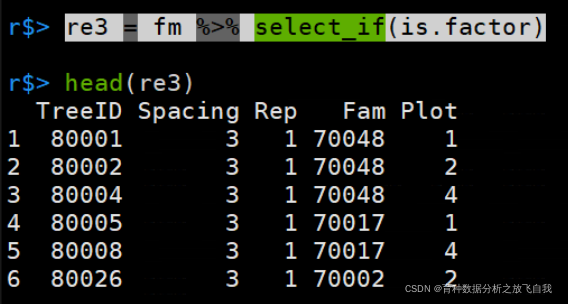
「匹配为因子的列:」
re3 = fm %>% select_if(is.factor)
同志们,你学会了吗?
关注我,不迷路。
❝欢迎关注我的公众号:
❞育种数据分析之放飞自我。主要分享R语言,Python,育种数据分析,生物统计,数量遗传学,混合线性模型,GWAS和GS相关的知识。
https://blog.sciencenet.cn/blog-2577109-1321918.html
上一篇:GCTA学习8 | 计算多性状遗传力和遗传相关
下一篇:BLUP育种值如何计算准确性