博文
氢气调控芳香烃受体,改善高氧暴露新生大鼠及RLE-6TN细胞的支气管肺发育不良
||
氢气调控芳香烃受体,改善高氧暴露新生大鼠及RLE-6TN细胞的支气管肺发育不良
前几天日本美国学者关于氢气分子机制研究中,发现氢气对线粒体复合物III中铁硫簇分子的作用,是非常有意义的研究。但在更多研究中,许多都会发现氢气对某种分子产生效应,但这些效应往往存在非排他性的问题。日本学者的研究也无法彻底回答分子靶点的细节过程,但研究中发现对复合物I没有作用具有非常重要的价值,因为从电子传递过程来说,复合物I地位更高,且也存在氢酶的基因后裔。都是氢化酶结构,有的有作用,有的没有,且不论其原因,这种差异就显得可贵了。看这个研究,更多是打靶类研究,虽然有负面确认,但没有说为什么找这种分子,因为这种分子在这种损伤中的作用,就显得过于生硬。存在的可能就是这种变化是病理过程特点,氢气只是因为对抗损伤,导致这种分子伴随发生。氢气医学研究中,这种情况的分子机制研究,非常多,非常多。
以上一段为本人点评
引言:本研究旨在探讨氢气(H₂)在高氧诱导支气管肺发育不良(bronchopulmonary dysplasia, BPD)中的作用及潜在机制,为开发有效的BPD治疗策略提供理论依据。
方法:构建高氧诱导的BPD大鼠模型及大鼠Ⅱ型肺泡上皮细胞(RLE-6TN)损伤模型。通过给予BPD大鼠氢气干预以评估其治疗效果,同时采用富氢培养基处理RLE-6TN细胞以检测细胞活力。通过体内外实验探究氢气对芳香烃受体(aryl hydrocarbon receptor, AHR)的调控作用,并通过AHR敲低及过表达实验明确AHR对细胞活力的影响。
结果:氢气治疗可改善BPD大鼠的肺组织病理损伤,减少细胞凋亡,上调表面活性物质蛋白SP-A和SP-B的表达,并调控AHR及其下游效应分子CPEB4的表达,进而减轻内质网(endoplasmic reticulum, ER)应激。体外实验中,富氢培养基可减轻RLE-6TN细胞损伤,促进AHR核转位,并激活CPEB4的表达;AHR过表达可提高RLE-6TN细胞活力,且AHR与CPEB4启动子区域具有强结合亲和力。
讨论:氢气通过调控AHR及其下游分子CPEB4,减轻内质网应激并减少细胞凋亡,从而改善高氧诱导的BPD。氢气的保护机制可能与调控AHR-CPEB4信号通路及缓解内质网应激密切相关。
1 引言
支气管肺发育不良(bronchopulmonary dysplasia, BPD)是一种慢性肺部疾病,其特征为肺血管发育受损、肺泡化停滞,进而导致肺功能下降。它仍是肺发育关键窗口期(孕24-38周)出生的极早产儿中最常见的并发症(1-4)。BPD的发病机制根本在于早产儿肺部固有的脆弱性。早产会中断囊状期(人类孕24-38周)的正常进程,阻碍其向肺泡期过渡——在此阶段,大多数肺泡通过分隔和微血管成熟形成(5)。这种发育中断导致早产儿肺部产生多种关键易感因素,使其易患BPD:
(i)结构脆弱性:出生时,早产儿肺部的气腔尚未发育成熟(为囊泡而非成熟肺泡),肺泡隔增厚,气体交换表面积减小,毛细血管网脆弱且未成熟(1-4)。这些结构缺陷使肺部难以适应宫外呼吸,也无法承受额外损伤。
(ii)抗氧化防御系统未成熟:早产儿体内关键抗氧化酶(包括超氧化物歧化酶、过氧化氢酶和谷胱甘肽)的水平显著降低(6, 7)。这种缺陷使发育中的肺部对氧化损伤高度敏感,即使在对足月儿安全的氧浓度下也不例外。
(iii)对氧毒性和炎症的敏感性升高:机械通气和补充氧气等挽救生命的干预措施虽至关重要,但也是BPD发病机制的主要诱因(8)。高氧暴露会超出未成熟的抗氧化能力(9),导致活性氧(reactive oxygen species, ROS)过量产生。ROS积累会损伤Ⅱ型肺泡上皮(alveolar epithelial type II, AT2)细胞(对表面活性物质合成和组织修复至关重要)和内皮细胞,引发强烈的肺部炎症(10)。氧化损伤与炎症损伤共同作用,破坏肺泡分隔和血管生成所需的信号通路。
(iv)修复能力受损:发育停滞、上皮和内皮损伤以及持续炎症共同损害了肺部的内在修复潜力(11-13)。由此产生的病理改变包括肺泡化停滞(肺泡结构简化、气腔扩大)、血管畸形,以及肺泡形成所需的上皮-间质相互作用紊乱。
因此,早产奠定了结构未成熟和抗氧化缺陷的病理基础。叠加高氧和机械通气的影响会加剧这种脆弱性(8, 9),最终导致BPD的典型特征——肺泡简化、血管化受损、AT2细胞耗竭和慢性炎症。这种发育未成熟与出生后氧化应激之间的协同作用,是使用高氧诱导新生大鼠模型(出生时处于类似囊状期)研究BPD发病机制和治疗方法的理论依据(14-16)。暴露于高氧环境的新生大鼠会再现人类BPD的形态学和分子特征,同时反映出与早产儿相似的遗传异质性(17)。
目前用于降低BPD风险的临床策略(如产前皮质类固醇、咖啡因、肺表面活性物质治疗和辅助药物)仅能带来有限益处(18, 19)。因此,迫切需要针对驱动疾病进展的氧化和炎症通路开发新型干预措施。氢气(H₂)作为一种具有强效抗氧化、抗炎和抗凋亡特性的治疗分子,已展现出良好前景(20-22)。多项研究证实其在慢性阻塞性肺疾病(COPD)和急性肺损伤(ALI)等呼吸系统疾病中的疗效(23)。近期研究发现,氢气吸入可通过抑制TLR4-NFκB-IL6/NLRP3信号通路、减少细胞因子和趋化因子产生,减轻胎盘炎症和脂多糖(LPS)诱导的BPD(6)。然而,氢气改善高氧诱导BPD的具体机制仍有待阐明。
芳香烃受体(aryl hydrocarbon receptor, AHR)是一种配体激活的转录因子,近期被证实是高氧暴露下发育中肺部氧化应激和炎症的关键调节因子。Shivanna等人的研究表明,新生Ahrd小鼠中AHR激活减少会加重高氧诱导的肺泡简化和炎症(24)。类似地,Rao等人报道,新生大鼠AHR缺乏会增加高氧条件下肺泡上皮细胞凋亡和血管损伤(25),凸显了该受体的保护作用。机制上,Bhattacharya等人发现,内源性配体犬尿氨酸激活AHR后,可上调核因子红系2相关因子2(Nrf2)和血红素氧合酶1(HO-1)等抗氧化酶,从而减轻早产肺模型中的氧化应激和肺泡损伤(26)。此外,Kuiper-Makris等人揭示,高氧通过AHR依赖的信号通路升高细胞内氧化应激并促进上皮-间质转化(EMT)(15)。体外补充研究表明,奥美拉唑诱导的AHR激活可抑制ROS产生和单核细胞趋化蛋白1(MCP-1)表达,减轻高氧诱导的人肺腺癌细胞(H441)损伤(27)。综上,来自动物、细胞和机制研究的证据表明,AHR是高氧诱导肺损伤的核心调节节点。其激活可显著抵御氧化和炎症损伤,使AHR信号通路成为BPD预防和治疗的潜在治疗靶点。
内质网(endoplasmic reticulum, ER)是负责蛋白质折叠、脂质合成和钙稳态的核心细胞器。它与其他细胞器保持密切的功能相互作用,对细胞内平衡至关重要。氧化应激、缺氧或营养缺乏引起的紊乱可诱导内质网应激并激活未折叠蛋白反应(UPR)。在BPD中,高氧驱动的氧化应激破坏内质网稳态,导致错误折叠蛋白(如表面活性物质蛋白SP-A和SP-B)积累,进而激活UPR(28, 29)。内质网应激与BPD发病机制密切相关:高氧会损害SP-A和SP-B的折叠与分泌,而这两种蛋白对肺泡稳定性至关重要。特别是SP-B下调已被证实与BPD中的肺泡发育不全和呼吸衰竭相关(30)。PERK和IRE1α等UPR传感器的激活,会通过CHOP介导的通路以及IL-6、TNF-α等促炎细胞因子的释放,进一步放大肺部炎症和凋亡,加重肺损伤(31)。Li等人证实,芒柄花素可通过AhR/CYP1A1通路抑制支气管上皮细胞的炎症、内质网应激和凋亡。Lee等人报道,AHR缺乏可减轻糖尿病肾病中氧化应激驱动的系膜细胞激活、巨噬细胞浸润和细胞外基质积累;而Guerrina等人发现,AhR缺乏会增加内质网应激敏感性(32-34)。另一项研究中,Feng等人发现,氢气吸入可通过减轻炎症、降低氧化应激和维持AhR蛋白水平,改善慢性低氧性肺动脉高压(CH-PH)大鼠的肺功能(35)。综上,这些研究结果表明,氢气可能通过减轻氧化应激缓解内质网应激,从而恢复SP-A/SP-B表达并调控AHR信号通路。
本研究旨在探讨氢气在高氧诱导BPD中的保护作用及潜在机制,以期发现促进肺生长和修复的新型治疗策略。
2 材料与方法
2.1 动物建模与干预
1周龄Sprague-Dawley(SD)大鼠(雌雄不限,体重15±3 g)购自广东致远生物医学科技有限公司。动物饲养于标准环境中,自由摄食饮水,维持12小时光照/12小时黑暗周期。每只哺乳母鼠喂养9只新生幼鼠,每个时间点设置12只幼鼠。所有动物实验均遵循ARRIVE指南(27)和英国《动物(科学程序)法案》(1986),并经来安科技(广州)有限公司实验动物伦理委员会批准(批准号:G2025039)。
为探究肺泡发育过程中的动态病理变化,选取三个时间点:第3天(早期损伤——氧化应激和凋亡)、第7天(肺泡化停滞——次级分隔受损)和第14天(BPD确立——持续性结构和功能缺陷)。新生大鼠及其代乳母鼠从出生后第0天起持续暴露于指定气体混合物中,直至第3、7或14天安乐死。大鼠通过二氧化碳(CO₂)窒息安乐死(流速为每分钟30%舱容置换,持续5分钟),随后通过颈椎脱臼确认死亡。
BPD模型通过新生大鼠出生后(D0)暴露于90%氧气诱导建立。为评估高氧和氢气的时间依赖性效应,将新生大鼠随机分为四组,并在三个出生后时间点(出生后第3、7和14天,以下简称D3、D7和D14)进行检测。每个时间点的分组如下:
(i)对照组(Con):幼鼠饲养于标准笼中,呼吸环境空气(21% O₂),不给予氢气;
(ii)氢气组(H₂):幼鼠饲养于密闭舱中,呼吸含2% H₂的21% O₂混合气体;
(iii)BPD组:幼鼠暴露于密闭舱中的90% O₂,诱导BPD;
(iv)BPD+氢气组(BPD+H₂):幼鼠暴露于密闭舱中含2% H₂的90% O₂混合气体。
每个时间点每组包含3只大鼠(n=3),总样本量N=36(4组×3时间点×3只大鼠)。该设计可动态评估高氧暴露和氢气干预过程中的肺病理变化、凋亡及表面活性物质蛋白表达。暴露舱内的气体浓度通过氢气检测仪(日本XP-3140型)和氧气检测仪(中国CY-12C型)持续监测。对照组幼鼠饲养于相同室内的标准笼中,环境条件维持在22-26℃,相对湿度60%-70%。
每天09:00打开舱门(时长<1小时),将母鼠在高氧组和常氧组幼鼠间轮换,以避免母鼠氧中毒,同时更换垫料并补充水和饲料。氢气由苏州摩尔气体设备有限公司设计制造的装置提供,可输出饱和浓度为2%的氢气。所有暴露实验均在透明密闭舱(长×宽×高:32×22×14 cm³)中进行,氢气浓度通过可燃气体检测仪(中国Aegisafe)实时监测。在第3、7和14天,通过CO₂吸入法对SD大鼠实施安乐死,通过无呼吸、无心跳和无反射确认死亡后,立即收集肺组织。右肺迅速冷冻于-80℃用于生化分析;左肺用于苏木精-伊红(HE)染色、免疫组织化学(IHC)染色和TUNEL染色,以确认BPD模型建立并评估氢气治疗的疗效。
2.2 酶联免疫吸附试验(ELISA)
在氢气治疗高氧诱导BPD模型的实验中,动物分为四组:对照组(Con)、氢气组(H₂)、BPD组(BPD)和BPD+氢气组(BPD+H₂)。第14天,对SD大鼠进行麻醉并采血,血液经1500 rpm离心10分钟后,收集血清,采用ELISA试剂盒检测丙氨酸转氨酶(ALT,Abcam ab234579)、天冬氨酸转氨酶(AST,Abcam ab263883)、肌酐(Crea,Mybiosource MBS749827)和尿素(Mybiosource MBS2600001)水平。
2.3 HE染色
在20 cm H₂O静水压下,通过气道向肺内缓慢灌注4%多聚甲醛。肺组织经石蜡包埋后,切成5 μm切片,采用苏木素染色液(中国ZLI-9610,zsbio)和水溶性伊红染色液(中国G1002,Servicebio)进行染色,随后在光学显微镜(NIKON ECLIPSE E100)下观察。形态学分析采用双盲设计:由独立研究人员对样本进行编号,分析人员未知实验组别信息。
为进行客观定量的形态学分析,本研究在40×物镜下对每张肺组织切片进行系统性图像采集和分析。为确保数据的代表性和客观性,在每张切片的非重叠、非连续区域随机选取6个独立视野,每个视野的实际面积为0.1 mm²,因此每张切片的总分析面积至少为0.6 mm²。所有图像分析均采用Image J软件进行,为维持分析条件的一致性,设置统一的阈值范围以精确区分目标结构与背景,所有测量工作由未知实验组别信息的研究人员完成,以最大限度减少主观偏差。其中,免疫组织化学染色的半定量分析采用积分光密度(IOD)值计算,同样基于上述标准化视野。
径向肺泡计数(RAC)和表面积(SA)的评估方法如前所述(36):RAC定义为从终末细支气管垂直延伸至最近胸膜的直线所穿过的闭合肺泡数量;肺泡隔厚度通过HE染色切片测量;肺泡化水平通过平均弦长(Lm)和肺泡表面积定量——Lm代表气腔壁之间的距离;校正40%的组织收缩率后,采用公式SA=4×组织体积密度(VDT)×肺体积/Lm×(1-0.4)计算肺泡表面积。
2.4 TUNEL染色
采用TUNEL染色试剂盒(GDP1042,中国Servicebio公司)检测肺组织中的细胞死亡数量,实验严格按照试剂盒说明书操作。通过明美显微镜数字成像系统(中国mshot公司)采集图像,细胞凋亡率以TUNEL阳性细胞面积与DAPI染色细胞核总面积的比值表示。每组设置3个肺组织样本,每个样本取3张切片计算平均值。
2.5 细胞培养与处理
大鼠Ⅱ型肺泡上皮细胞(RLE-6TN,CRL-2300,ATCC)购自美国ATCC细胞库(https://www.atcc.org/),使用经认证的细胞材料进行实验。RLE-6TN细胞接种于含10%胎牛血清(FBS,A5670701,美国Gibco公司)和1%青霉素-链霉素溶液(100 μg/ml,15070063,美国Gibco公司)的Ham's F-12K培养基(21127022,美国Gibco公司)中,置于37℃、5% CO₂培养箱中培养。随后将细胞随机分为四组:对照组(Con)、氢气组(HRM)、高氧组(Hyperoxia)和高氧+氢气组(Hyperoxia+HRM)。
富氢培养基(HRM)参照先前描述的方法[38]制备:在0.4 MPa压力下将H₂溶解于Ham's F-12K培养基中2小时,达到过饱和状态,最终氢气浓度稳定在0.6 mmol/L。采用亚甲基蓝-氧化还原滴定法检测完全培养基中的氢气浓度:取5 ml含氢培养基样本,用亚甲基蓝胶体铂(MB-Pt)试剂滴定至蓝色恰好不再消失,即为滴定终点。理论上,每滴胶体铂MB-Pt试剂对应0.1 mmol/L的氢气浓度(例如:加入8滴试剂时,未显色的氢气浓度为0.8 mmol/L;加入12滴时为1.2 mmol/L),持续滴定至含氢培养基恰好变蓝。含氢溶液中氢气浓度的计算方法为:加入的胶体铂MB-Pt试剂滴数除以10。
需进行高氧处理的细胞,将5% CO₂与95% O₂的混合气体通入氧培养箱后,置于培养箱中孵育。不同细胞组分的蛋白提取实验中,各组(Con、HRM、Hyperoxia、Hyperoxia+HRM)RLE-6TN细胞经预冷磷酸盐缓冲液(PBS)洗涤2次后收集,采用核质蛋白提取试剂盒(P0027,中国Beyotime公司)提取核蛋白与细胞质蛋白:细胞经PBS洗涤后离心收集沉淀;提取细胞质蛋白时,向沉淀中加入适量细胞质提取试剂,充分涡旋使细胞完全裂解后离心,立即将上清液(含细胞质蛋白)转移至预冷管中;提取核蛋白时,先彻底吸弃上述离心后的残留上清液以避免细胞质污染,向沉淀中加入适量核提取试剂,间隔剧烈涡旋使沉淀充分分散,后续离心后立即将上清液(含核蛋白)转移至新的预冷管中。
AHR配体FICZ干预高氧处理RLE-6TN细胞的实验中,细胞分为四组:对照组(Con)、FICZ组(FICZ)、高氧组(Hyperoxia)和高氧+FICZ组(Hyperoxia+FICZ)。高氧处理方法同前所述,FICZ(SML1489,美国Sigma公司)使用浓度为10 μmol/L。
2.6 流式细胞术
检测SP-A和SP-B的流式细胞术实验中,通过气管向肺组织内注入中性蛋白酶(dispase),并将肺组织置于中性蛋白酶和Ⅳ型胶原酶中,37℃孵育40分钟,期间频繁振荡。消化后的组织依次通过100 μm、70 μm和40 μm细胞筛网,获得单细胞悬液。细胞重悬于流式细胞术缓冲液中,浓度调整为5×10⁶个/ml。采用荧光激活细胞分选(FACS)分离AT2细胞,分选表型为:PI⁻(碘化丙啶阴性,排除死细胞)、CD45⁻(泛造血细胞标志物CD45阴性,排除免疫细胞)、CD31⁻(内皮细胞标志物CD31阴性,排除内皮细胞)、EpCAM⁺(上皮细胞黏附分子EpCAM阳性,富集上皮细胞)、LysoTracker⁺(溶酶体追踪剂阳性,该荧光染料可在AT2细胞特征性的板层小体中积累)。
RLE-6TN细胞经4%多聚甲醛室温固定30分钟后,用10%山羊血清室温封闭1小时,加入相应一抗4℃孵育过夜,随后加入合适的二抗室温孵育30分钟,采用流式细胞仪(Accuri™,美国BD Biosciences公司)检测,每个样本记录10,000个事件用于分析。本研究使用的抗体包括:SP-A(1:500,11850-1-AP,中国Proteintech公司)、SP-B(1:500,1034R,中国Yajimall公司)、山羊抗兔IgG H&L(Alexa Fluor® 488)(ab150077,英国Abcam公司)、山羊抗小鼠IgG H&L(Alexa Fluor® 488)(ab150113,英国Abcam公司)。细胞凋亡率检测采用Annexin V-FITC/PI凋亡检测试剂盒(G1511,中国Solarbio公司),按照说明书操作并通过流式细胞仪分析。
2.7 免疫组织化学(IHC)染色
肺组织石蜡切片经脱蜡处理后,置于3%过氧化氢溶液中孵育10分钟;室温下用10%山羊血清封闭10分钟,随后加入一抗37℃孵育2小时;加入相应二抗37℃孵育30分钟,PBS洗涤3次后,置于辣根过氧化物酶标记的链霉亲和素工作液中37℃孵育5分钟;采用DAB显色剂显色10分钟,洗涤后树脂封片,通过明美显微镜数字成像系统(中国mshot公司)拍照。采用积分光密度(IOD)量化染色强度,评估p-AHR、AHR和CPEB4的表达水平。每张肺切片的形态学分析面积至少为0.6 mm²(40×物镜下随机选取6个非重叠视野,每个视野面积为0.1 mm²),使用Image J软件在一致的阈值设定下计算IOD值,该值代表染色区域的总光密度,反映样本中抗原的表达水平。本研究使用的抗体包括:p-AHR(1:1000,PA5-104880,美国Thermo Fisher公司)、AHR(1:800,67785-1-IG,中国Proteintech公司)、CPEB4(1:250,25342-1-AP,中国Proteintech公司)、山羊抗兔IgG H&L(HRP)(1:1000,GB23303,中国Servicebio公司)、山羊抗小鼠IgG/HRP(1:1000,GB23301,中国Servicebio公司)。
2.8 蛋白质印迹(WB)分析
采用核质蛋白提取试剂盒(P0027,中国Beyotime公司)提取核蛋白与细胞质蛋白;组织和细胞总蛋白采用预冷的RIPA裂解液(强配方,Beyotime公司,P0013B)提取。为抑制蛋白降解和去磷酸化,向裂解液中加入1 mmol/L PMSF(Beyotime公司,ST506)以及蛋白酶和磷酸酶抑制剂混合物(Roche公司,04693132001和04906837001)。裂解后,组织样本于4℃、12,000 rpm离心15分钟,收集上清液作为总蛋白提取物。采用BCA蛋白定量试剂盒(Beyotime公司,P0012)精确测定蛋白浓度。
为确保蛋白质印迹结果的可靠性和可比性,计算后标准化每孔总蛋白上样量:组织来源样本为30 μg,细胞来源样本为20 μg。该上样量经预实验验证,确保目标条带的信号强度处于线性范围内且未过饱和。所有样本与5×SDS-PAGE上样缓冲液混合,100℃煮沸10分钟使蛋白完全变性后进行电泳。电泳结束后,将蛋白转移至PVDF膜上,用5% BSA封闭1小时;加入相应一抗4℃孵育过夜,随后加入对应二抗室温孵育2小时;采用增强化学发光(ECL)法显色,通过自动凝胶成像分析仪定量条带灰度值。
本研究使用的抗体包括:p-AHR(1:1000,PA5-104880,美国Thermo Fisher公司)、AHR(1:1000,MA1-513,美国Thermo Fisher公司)、CPEB4(1:1000,PA5-25538,美国Thermo Fisher公司)、p-IRE1α(1:1000,PA5-105424,美国Thermo Fisher公司)、IRE1α(1:500,MA5-14991,美国Thermo Fisher公司)、GAPDH(1:10000,ab8245,英国Abcam公司)、β-肌动蛋白(β-actin,1:10000,ab8226,英国Abcam公司)、组蛋白(histone,1:10000,ab1791,英国Abcam公司)、山羊抗兔IgG H&L HRP(1:10000,ab6721,英国Abcam公司)、山羊抗小鼠IgG/HRP(1:5000,SE131,中国Solarbio公司)。使用ImageJ软件将p-AHR的条带强度以AHR为内参进行标准化,其他所有蛋白条带强度均以GAPDH为内参进行标准化。
2.9 AHR敲低与过表达
小干扰RNA阴性对照(si-NC)、AHR小干扰RNA(si-AHR)、空载体(vector)和AHR过表达载体(oe-AHR)序列由北京擎科生物科技有限公司合成。按照制造商说明,使用Lipofectamine 3000(Invitrogen公司,美国)转染小干扰RNA,转染48小时后收集细胞。详细序列见补充材料数据1-3。
2.10 细胞活力评估
采用M5 HiPer细胞计数试剂盒(MF128-01,中国Mei5bio公司)评估细胞活力。细胞以3×10³个/孔的密度接种于96孔板中,接种后24、48和72小时,向每孔加入10 μl CCK-8试剂,37℃避光孵育2小时,随后使用酶标仪(ELX800,美国BioTek公司)测定450 nm处的吸光度值。
2.11 实时定量聚合酶链反应(qPCR)
采用Trizol溶液裂解细胞,通过氯仿和异丙醇提取RNA。按照M5 HiPer第一链cDNA合成试剂盒(MF011-01,中国Mei5bio公司)的说明书进行mRNA逆转录,所得cDNA用于qPCR实验。实验流程按照PerfectStart® Green qPCR SuperMix试剂盒(MF013,中国Mei5bio公司)操作。使用的引物序列如下:AHR(上游:CACAGAGACCGGCTGAACAC,下游:TGCTGAAAGCCCAGGTAATCT);CPEB4(上游:ACGGGTTTGGAGTGCTAGTG,下游:CCCCTGGATTTTCTTCGGCT);GAPDH(上游:AATGACCCCTTCATTGAC,下游:TCCACGACGTACTCAGCGC)。
2.12 染色质免疫沉淀-实时定量聚合酶链反应(ChIP-qPCR)
按照ChIP检测试剂盒(ab156907,英国Abcam公司)的说明书进行实验。细胞经37%甲醛交联后,用125 mmol/L甘氨酸处理;裂解并超声破碎后,将消化后的染色质与抗体混合,4℃振荡孵育过夜;随后向免疫沉淀反应体系中加入磁珠,4℃振荡混合1小时;洗脱免疫沉淀的染色质DNA,通过qPCR定量。使用的抗体包括抗AHR抗体(ab2769,英国Abcam公司)和IgG同型对照抗体(3900,美国Cell Signaling Technology公司)。详细引物序列见补充材料数据4-5。
2.13 免疫荧光染色
RLE-6TN细胞经4%多聚甲醛固定30分钟,0.5%曲拉通(9002-93-1,中国Solarbio公司)透化10分钟,26℃血清封闭1小时;加入CPEB4抗体(1:250,25342-1-AP,中国Proteintech公司)和AHR抗体(1:800,67785-1-IG,中国Proteintech公司)4℃孵育过夜,随后加入荧光二抗,26℃孵育2小时;DAPI染色细胞核,通过激光共聚焦显微镜(LSM 900,德国ZEISS公司)观察荧光强度,使用Image J软件定量免疫阳性细胞。
2.14 统计学分析
所有实验均采用独立生物学重复样本进行三次重复,数据以平均值±标准差表示。采用Shapiro-Wilk检验验证数据正态性,Levene检验评估方差齐性。多组间比较采用单因素方差分析(one-way ANOVA),若整体差异有统计学意义(P<0.05),则进行最小显著差异法(LSD)事后检验。采用四分位距(IQR)法识别异常值,排除低于Q₁-1.5×IQR或高于Q₃+1.5×IQR的值。所有统计学分析使用GraphPad Prism 9.0软件(GraphPad Software公司,美国加利福尼亚州拉霍亚)进行,P<0.05认为差异具有统计学意义。
3 结果
3.1 氢气改善高氧诱导的支气管肺发育不良(BPD)
采用苏木精-伊红(HE)染色评估大鼠肺组织的病理改变。如图1A-C所示,第3天(病理早期),BPD组出现肺泡隔轻度增厚、肺泡分支密度降低和径向肺泡计数(RAC)轻微下降,但这些变化无统计学意义;肺泡总数与平均单个肺泡表面积的乘积(总肺泡表面积)无明显改变;RAC的轻微降低提示平均线性截距(Lm)代偿性增加,反映肺泡发育损伤的起始。
第7天(损伤进展期),对照组肺泡持续成熟,肺泡数量随正常发育增加;而BPD组肺泡数量显著减少、气腔扩大,RAC明显下降(P<0.05),Lm同时增加(肺泡化延迟的典型特征),平均单个肺泡表面积也显著增大(P<0.05)。此阶段,肺泡数量的减少超过了肺泡扩大的代偿作用,结合肺泡隔增厚和结构紊乱,导致总肺泡表面积显著下降,提示肺泡结构简化。
第14天,对照组肺泡排列紧密均匀,而BPD组肺泡明显扩大,RAC显著降低(P<0.05),Lm增加,平均肺泡表面积升高(P<0.05),总肺泡表面积进一步下降,证实肺泡结构简化。相比之下,氢气处理组的上述病理特征显著减轻(P<0.05),肺泡形态接近对照组,RAC恢复(逆转BPD诱导的降低),平均肺泡表面积减小。
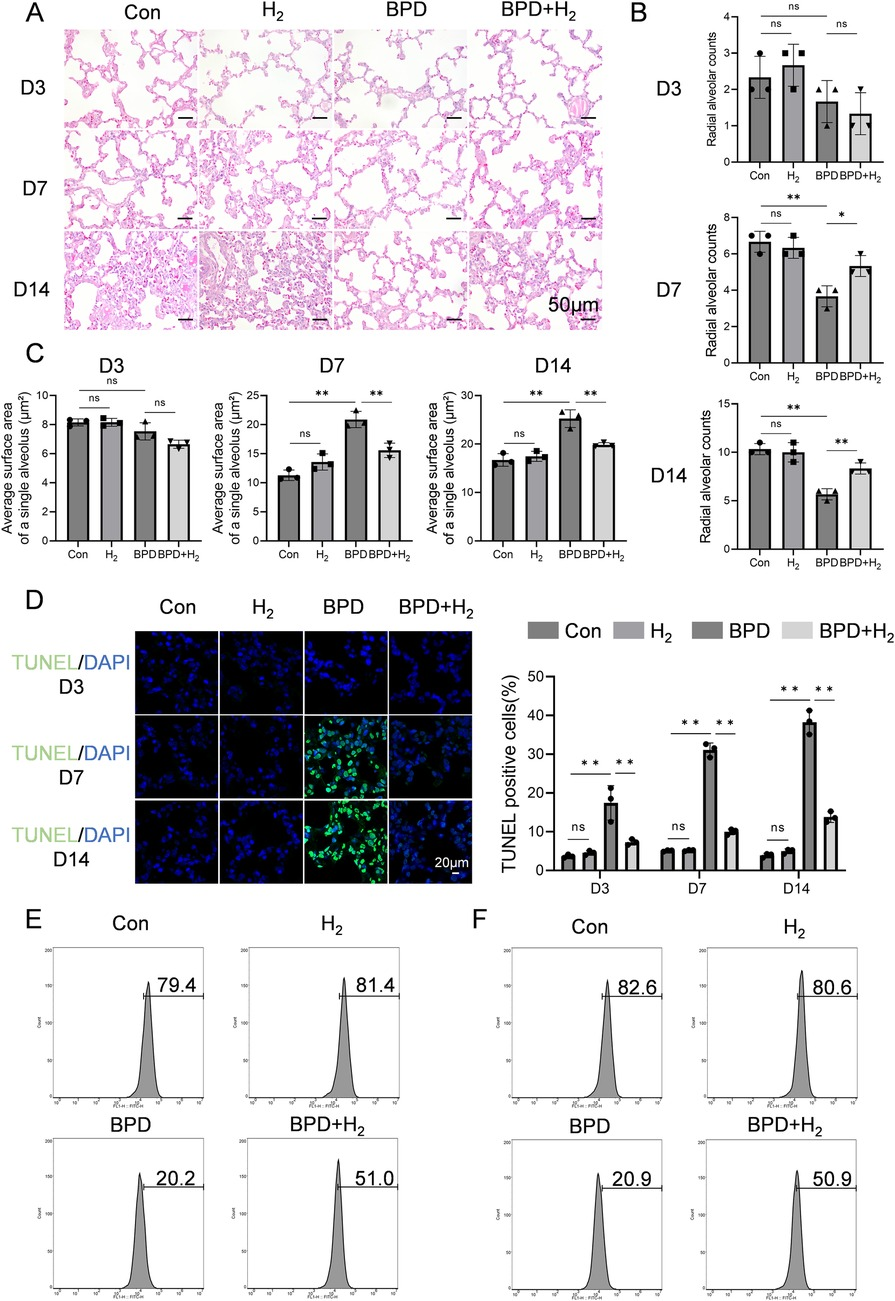
图1 氢气改善高氧诱导的支气管肺发育不良(BPD)
(A) 苏木精-伊红(HE)染色评估BPD大鼠肺组织病理状态,20倍放大,比例尺=50μm。(B) 径向肺泡计数(RAC)分析,AHR激活形式(磷酸化形式)主要富集于肺泡上皮细胞核内,而CPEB4在细胞质及核周区域高表达。(C) 单个肺泡平均表面积定量分析(μm²)。(D) TUNEL染色检测肺组织凋亡情况。(E) 流式细胞术检测对照组(Con)、氢气组(H₂)、BPD组及BPD+氢气组大鼠第14天肺组织中SP-A的表达水平。(F) 流式细胞术检测上述四组大鼠第14天肺组织中SP-B的表达水平。数据以平均值±标准差表示(n=3)。
如图1D所示,通过TUNEL染色评估BPD模型大鼠肺泡上皮细胞凋亡情况,并定量分析TUNEL阳性细胞比例以反映凋亡强度。随时间推移(第3天→第7天→第14天),BPD组细胞凋亡呈现“起始→峰值→持续”的变化模式,其中第7天为凋亡高峰,与HE染色观察到的中期结构损伤最显著阶段一致。第3天时,对照组仅观察到微弱绿色荧光,提示正常发育状态;而BPD组荧光强度略有增加,表明凋亡启动及损伤诱导的细胞死亡开始发生;BPD+氢气组荧光强度介于两者之间,提示早期氢气干预可部分抑制凋亡。第7天时,BPD组出现强绿色荧光,提示广泛凋亡发生;而BPD+氢气组荧光强度显著降低(P<0.05),接近对照组水平,表明氢气可有效抑制细胞凋亡。第14天时,BPD组持续存在绿色荧光,提示细胞死亡持续发生且修复不完全;而BPD+氢气组荧光强度显著减弱,表明氢气具有持续的抗凋亡和细胞保护作用。
流式细胞术检测表面活性物质蛋白SP-A和SP-B的表达结果显示,与BPD模型组相比,氢气处理组表达SP-A和SP-B的Ⅱ型肺泡上皮(AT2)细胞比例显著升高(P<0.05;图1E、F)。这些发现表明,氢气治疗可在高氧应激下维持AT2细胞完整性,保留其合成表面活性物质蛋白的功能。同时,对各组(对照组、氢气组、BPD组、BPD+氢气组)血清生化指标[丙氨酸转氨酶(ALT)、天冬氨酸转氨酶(AST)、肌酐(CREA)、尿素(UREA)]的检测结果显示,组间无统计学显著差异(P>0.05;补充图S1),表明氢气治疗未引发全身毒性。
3.2 氢气调控AHR和CPEB4,减轻内质网应激
基于染色质免疫沉淀测序(ChIP-seq)数据(ENCODE数据库)的生物信息学预测显示,芳香烃受体(AHR)可直接结合CPEB4启动子区域,该区域位于转录起始位点(TSS)上游482bp处(图2A)。p-AHR、总AHR及CPEB4的免疫组织化学染色结果显示,AHR/p-AHR特异性定位于肺泡上皮细胞和间质细胞的细胞核内,CPEB4定位于细胞质内,与预期的亚细胞分布一致(图2B)。采用积分光密度(IOD)对染色强度进行定量分析发现,BPD组p-AHR的IOD值显著低于对照组(P<0.01;图2C),而总AHR无显著组间差异(图2D);同理,BPD组CPEB4的IOD值显著降低(P<0.01;图2E)。值得注意的是,与BPD组相比,BPD+氢气组的p-AHR和CPEB4 IOD值均显著升高(P<0.01),表明氢气治疗可恢复二者的表达水平。
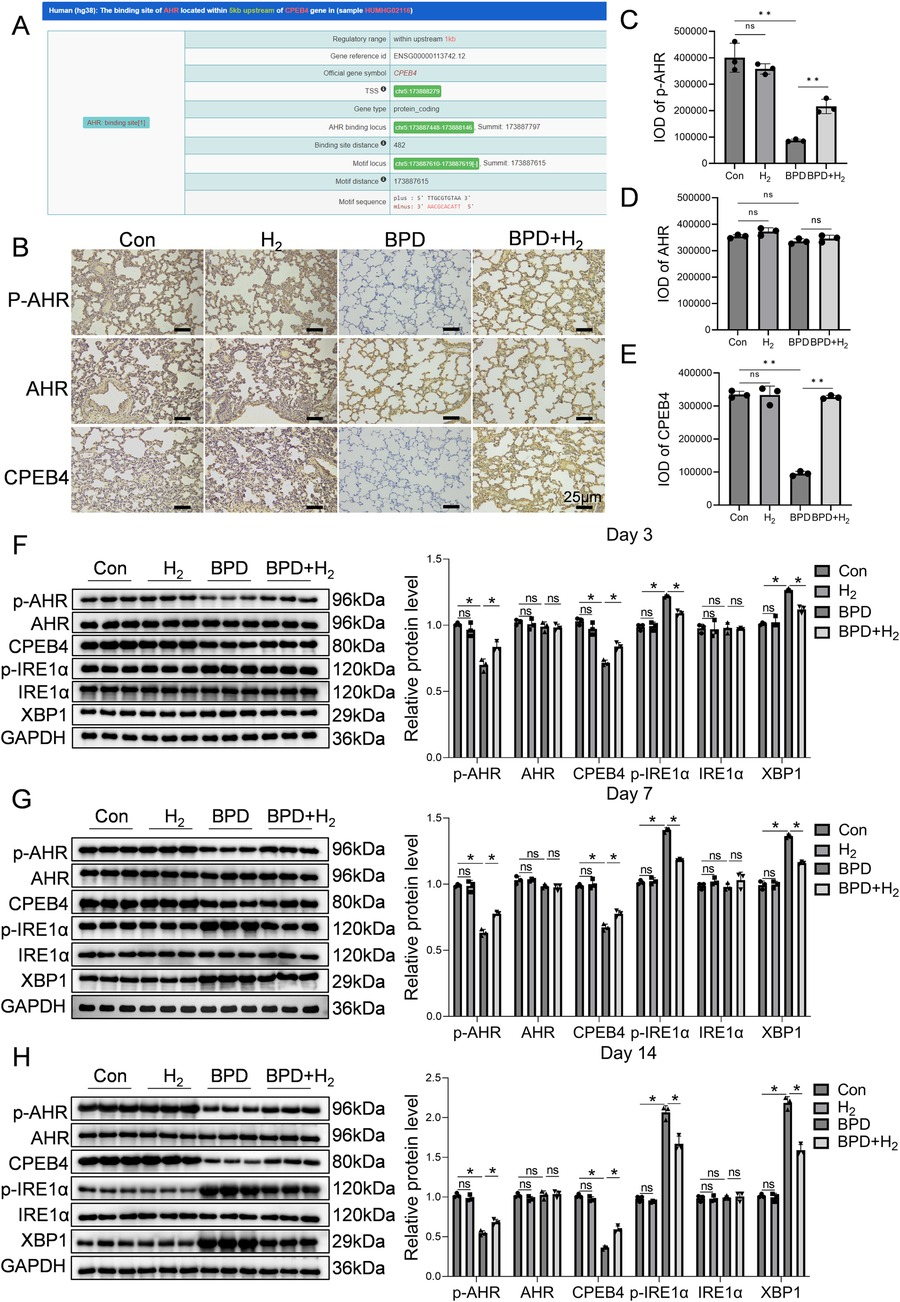
图2 氢气激活AHR及其下游分子CPEB4,进而减轻内质网应激
(A) 展示AHR的预测下游调控分子。(B) 免疫组织化学检测肺组织中AHR激活状态及CPEB4表达水平,60倍放大,比例尺=25μm,AHR激活形式(磷酸化形式)主要富集于肺泡上皮细胞核内,而CPEB4在细胞质及核周区域高表达。(C-E) p-AHR、AHR及CPEB4蛋白表达的IOD定量分析。(F-H) 蛋白质印迹法检测第3、7、14天p-AHR、AHR、CPEB4、p-IRE1α、IRE1α及XBP1的蛋白表达水平,并对蛋白条带进行定量分析。数据以平均值±标准差表示(n=3),*P<0.05。
蛋白质印迹分析结果验证了上述发现(图2F-H):BPD组p-AHR水平显著降低(P<0.05),而BPD+氢气组恢复至接近对照组水平,总AHR无明显变化;相反,BPD组磷酸化IRE1α(p-IRE1α)水平升高(P<0.05),BPD+氢气组则降低并恢复至对照组水平,总IRE1α无变化,提示BPD中IRE1α通路过度激活;同理,BPD组CPEB4表达显著降低(P<0.05),经氢气治疗后恢复;BPD组XBP1水平升高(P<0.05),氢气治疗可抑制其表达。综上,这些结果表明,氢气可恢复CPEB4表达,抑制p-IRE1α过度激活,减弱下游XBP1信号传导,从而减轻BPD中内质网应激相关损伤。
3.3 氢气减轻RLE-6TN细胞损伤
采用CCK-8法评估RLE-6TN细胞活力,结果如图3A所示,与常氧对照组(Con组)相比,高氧暴露72h后细胞活力显著降低(P<0.05),证实高氧可诱导肺泡上皮细胞损伤和死亡;而联合氢气处理[高氧+富氢培养基(HRM)组]可显著改善细胞活力,吸光度值显著高于高氧组(P<0.05),接近常氧对照组水平。流式细胞术进一步证实,高氧可增加RLE-6TN细胞凋亡率,而HRM处理可显著降低凋亡(图3B)。实时定量聚合酶链反应(qPCR)结果显示,高氧可降低AHR和CPEB4的mRNA表达,HRM处理可恢复二者表达(图3C)。蛋白质印迹分析显示,高氧可抑制AHR磷酸化,下调CPEB4表达,激活IRE1α,增加XBP1表达;相反,HRM处理可增强AHR磷酸化,上调CPEB4表达,抑制IRE1α激活,减少XBP1表达(图3D)。为进一步探究AHR定位,免疫荧光分析显示,常氧条件下AHR主要定位于细胞核内,而高氧可诱导其向细胞质转位(图3E);值得注意的是,高氧暴露后细胞质AHR水平显著升高,而HRM处理可促进AHR向细胞核显著重新分布(补充图S2)。这些发现表明,HRM可在高氧应激下促进AHR核转位。
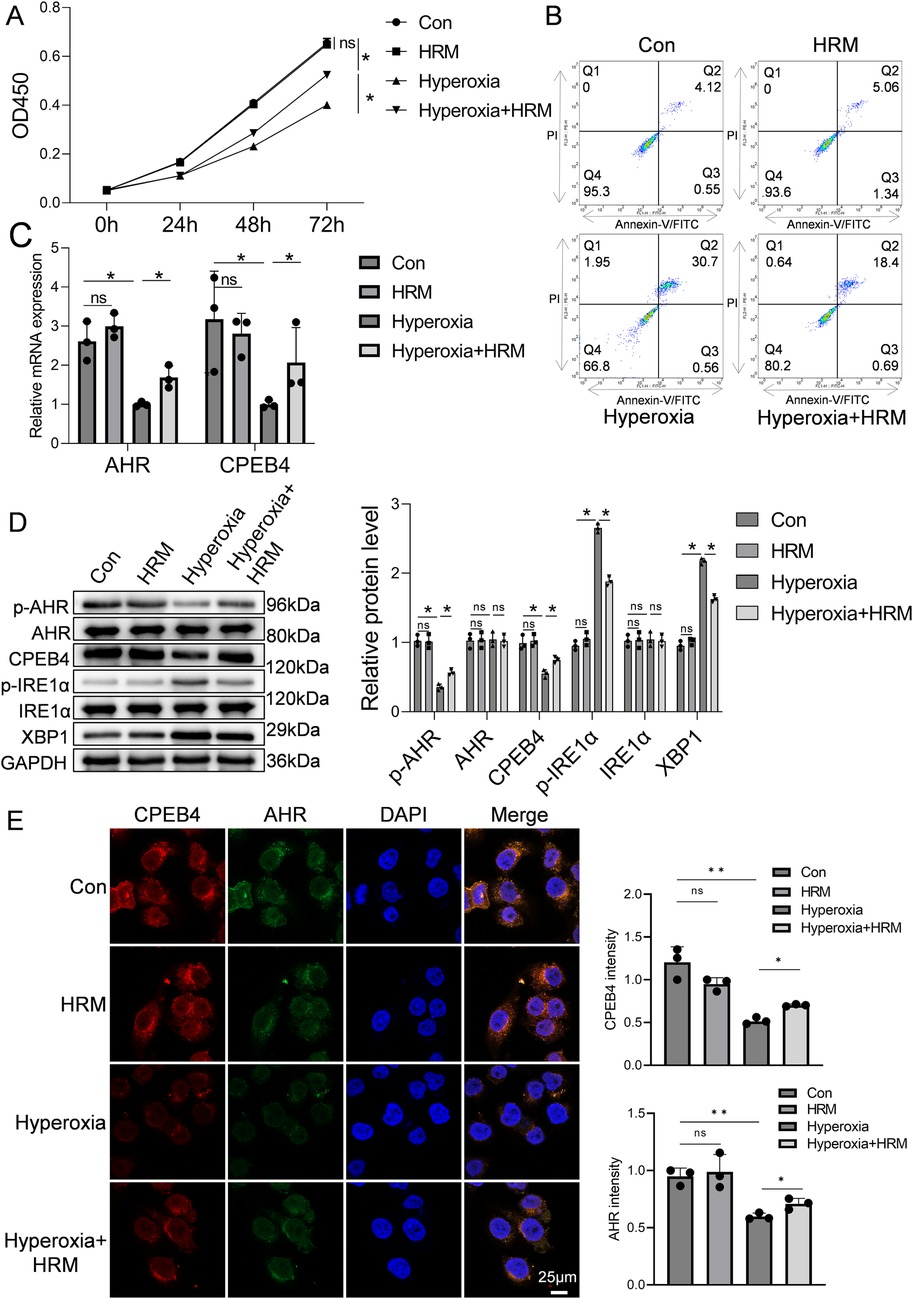
图3 探究氢气在高氧环境中对RLE-6TN细胞的影响
(A) CCK-8法评估RLE-6TN细胞活力。(B) 流式细胞术进一步评估RLE-6TN细胞凋亡情况。(C) qPCR检测RLE-6TN细胞中AHR和CPEB4的mRNA水平。(D) 蛋白质印迹法分析RLE-6TN细胞中p-AHR、AHR、CPEB4、p-IRE1α、IRE1α及XBP1的表达水平,并对蛋白条带进行定量分析。(E) 免疫荧光显微镜观察RLE-6TN细胞中AHR的核转位情况,60倍放大,比例尺=25μm。数据以平均值±标准差表示(n=3),*P<0.05。
3.4 AHR促进RLE-6TN细胞活力
对高氧处理的RLE-6TN细胞给予AHR激动剂FICZ干预,结果如图4A所示,高氧显著降低细胞活力,而FICZ处理可显著恢复细胞活力;同理,高氧下细胞凋亡率升高,FICZ暴露后凋亡率显著降低(图4B)。qPCR结果显示,高氧抑制AHR mRNA表达,FICZ可逆转该效应(图4C)。蛋白质印迹分析显示,高氧抑制AHR磷酸化,下调CPEB4,激活IRE1α,升高XBP1表达;相反,FICZ处理可增强AHR磷酸化,上调CPEB4,抑制IRE1α激活,降低XBP1表达(补充图S3)。
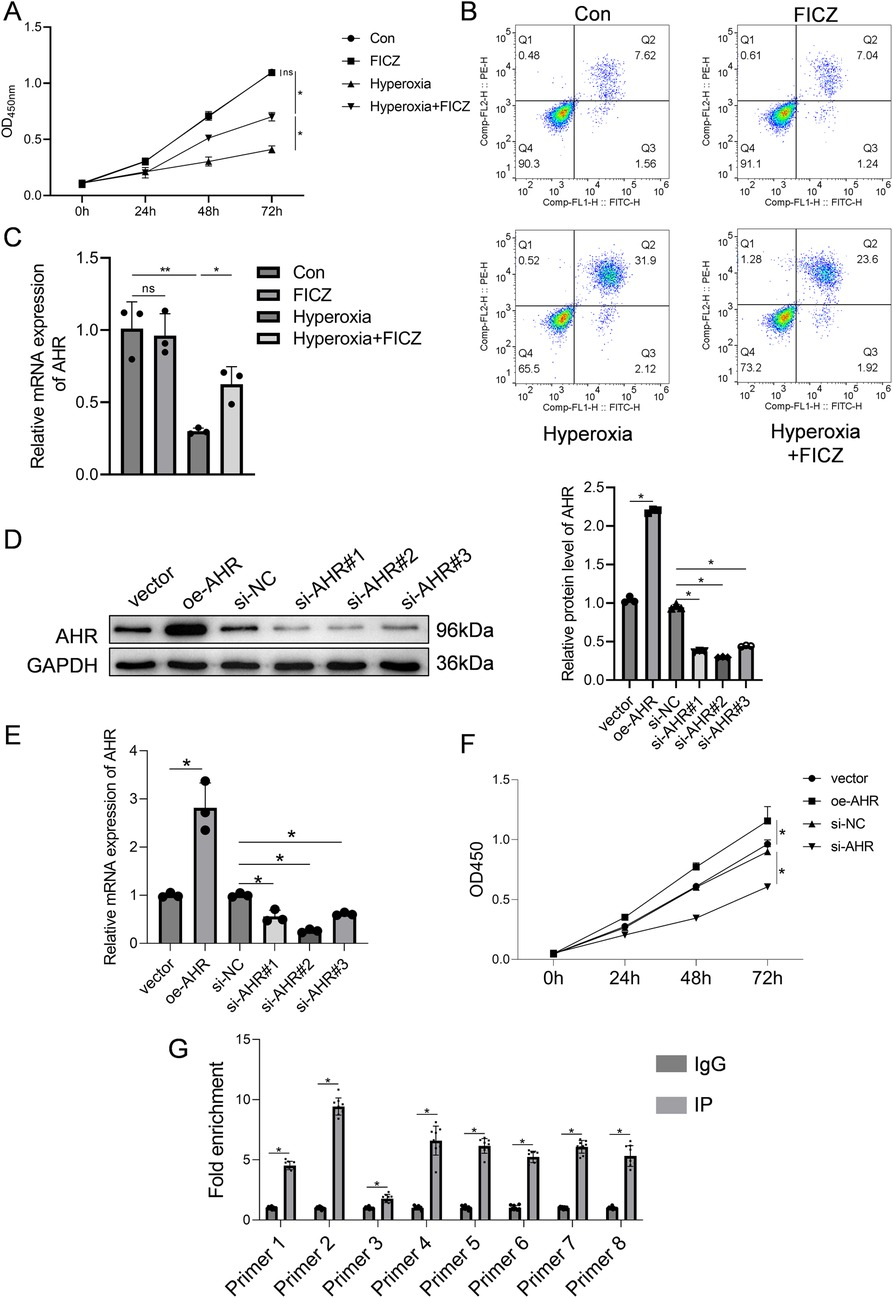
图4 探究AHR在高氧环境中对RLE-6TN细胞的影响
(A) RLE-6TN细胞活力检测结果。(B) RLE-6TN细胞凋亡检测结果。(C) RLE-6TN细胞中AHR的mRNA表达水平。(D) 蛋白质印迹法验证RLE-6TN细胞中AHR过表达及敲低效率。(E) qPCR验证RLE-6TN细胞中AHR过表达及敲低效率。(F) CCK-8法评估AHR过表达及敲低后RLE-6TN细胞的活力。(G) ChIP-qPCR检测AHR与CPEB4启动子的结合情况(3次独立实验,共9个生物学重复)。数据以平均值±标准差表示,A-F组n=3,*P<0.05。
通过qPCR和蛋白质印迹验证AHR敲低及过表达效率:过表达组AHR蛋白和mRNA水平分别是空载体对照组的2.2倍和2.8倍(图4D、E),证实过表达成功;在测试的3条小干扰RNA(siRNA)中,si-AHR2敲低效果最佳,AHR蛋白和mRNA水平分别降至si-NC组的0.3倍和0.26倍,因此选择该序列用于后续实验(图4D、E)。功能学实验显示,AHR敲低可抑制RLE-6TN细胞活力,而AHR过表达可显著增强细胞活力(图4F)。为明确AHR是否直接结合CPEB4启动子,ChIP-qPCR分析证实AHR与CPEB4启动子区域具有强结合亲和力(图4G)。
4 讨论
本研究采用成熟的高氧诱导新生大鼠BPD模型,该模型在可重复的实验条件下能再现“新型BPD”的典型特征,已得到广泛认可(17, 37)。干预措施未影响肝肾功能,所有动物均表现出良好的存活率。HE染色结果显示,与对照组相比,高氧组肺泡结构简化、肺泡数量减少、气腔扩大、肺泡隔增厚,证实BPD模型构建成功。鉴于AHR在肺、胎盘等氧交换组织中高表达(38),且AHR信号通路在BPD发病机制中具有重要作用(26),本研究进一步探究了其调控功能。蛋白质印迹分析显示,高氧抑制AHR磷酸化,而氢气治疗可恢复其磷酸化水平并减轻BPD病理损伤。先前研究表明,AHR激活可保护新生小鼠肺组织和人胎儿肺微血管内皮细胞免受高氧损伤(39),且药物激活AHR可减轻早产儿外周血单核细胞的氧化应激(40)。
本研究进一步发现,氢气可促进AHR核转位,揭示了一种新的调控机制。生理条件下,AHR主要定位于细胞质,核转位后获得转录活性(41)。染色质免疫沉淀实验证实AHR与CPEB4启动子存在强结合作用。高氧抑制AHR磷酸化和CPEB4表达,提示氧化应激诱导的内质网应激参与翻译失调(28)。由于CPEB4调控肺泡上皮细胞增殖和凋亡相关mRNA的翻译,其下调会破坏翻译稳态,促进凋亡和肺泡损伤,这与TUNEL和HE染色结果一致。氢气可恢复CPEB4表达,稳定翻译平衡,并通过降低IRE1α磷酸化和XBP1表达减轻内质网应激。IRE1α作为未折叠蛋白反应(UPR)的核心传感器,在内质网应激时激活并剪切XBP1 mRNA,进而启动适应性修复或凋亡信号(42)。与先前在新生BPD模型中观察到的内质网应激升高结果一致(32, 43),本研究在BPD大鼠肺组织和高氧暴露的RLE-6TN细胞中均检测到IRE1α激活增强和XBP1水平升高,凸显内质网应激是BPD的治疗靶点。
基于氢气的抗氧化和细胞保护特性,本研究探究了其在高氧诱导BPD中的治疗效果。临床前证据证实,2%-4%氢气安全、易扩散,可有效中和活性氧(ROS)且不影响氧合(44)。本研究采用定制气体输送系统生成2%氢气-氧气混合物,该浓度兼具疗效和安全性:浓度>4%具有可燃性,而<1%则无治疗效果(45)。根据亨利定律,吸入2%氢气可使血浆浓度达到约0.4-0.6mmol/L(46),亚甲基蓝滴定法证实富氢培养基(HRM,0.6mmol/L)可维持稳定氢气水平达24h(47)。研究结果表明,氢气通过调控AHR-CPEB4-内质网应激轴、增强表面活性物质合成,改善高氧诱导的BPD。SP-A和SP-B对降低肺泡表面张力(48)、维持肺弹性至关重要,其在BPD大鼠中显著降低,而氢气治疗可恢复其表达。在RLE-6TN细胞中,氢气通过直接结合启动子区域增强AHR磷酸化和核转位,上调CPEB4表达,改善细胞活力并减少凋亡。这些效应与AHR激动剂FICZ的作用一致,且可被AHR敲低所阻断,证实AHR在氢气介导的高氧应激细胞保护中具有关键作用。
综上,本研究揭示了一种新机制:氢气通过激活AHR上调CPEB4表达,从而抑制内质网应激、增强表面活性物质蛋白合成,改善新生大鼠BPD的肺功能和结构。然而,本研究仍存在一些局限性:RLE-6TN细胞源自成年大鼠肺泡上皮,可能无法完全再现新生肺的未成熟表型,未来研究将采用出生后1-3天大鼠的原代肺泡上皮细胞,以更好地反映发育生物学特征;体内实验需延长观察周期,评估体重、潮气量、呼吸频率、气道阻力等功能指标,以及肺泡简化、血管重塑、炎症等结构病理参数;需采用液相色谱-质谱法(LC-MS)定量生物基质中的氢气水平,明确剂量-效应关系;此外,石蜡包埋可能导致不可预测的组织收缩(49),需结合补充形态测量方法以提高准确性;最后,检测AHR拮抗剂是否可阻断氢气的保护作用,对确认因果关系至关重要。体内外凋亡反应的差异(可能源于体内特有的代偿性抗氧化机制)也强调了需要肺类器官等生理相关研究平台。
5 结论
综上,本研究结果表明,氢气通过激活AHR-CPEB4通路减轻内质网应激和细胞凋亡,为高氧诱导的BPD提供了一种有前景的治疗策略。
https://blog.sciencenet.cn/blog-41174-1512882.html
上一篇:首个慢性疲劳综合征血液检测方法问世
下一篇:星形胶质细胞塑造人类行为、记忆与健康
