博文
顶刊速报|山东师范孙旭平教授、华中师范朱成周教授、中科大焦淑红教授等电解水催化剂最新成果速览
||
引言
随着全球环境问题的日益凸显,人类对清洁能源的需求越来越大,其中氢能作为一种能量密度高、清洁的可再生能源,将成为石油、煤炭等传统化石燃料的理想替代能源。因此,发展氢能经济对于构建低碳能源体系、实现“碳达峰”及“碳中和”目标具有重要意义。目前,氢能主要通过化石能源的分解来制备,但这不仅会消耗大量的不可再生能源,而且还会产生温室气体。相反,电解水制氢技术由于具有操作简单、副反应少、绿色经济等特点成为了未来重要的制氢方式之一。为了降低反应能耗,通常需要开发高活性、高稳定性的阳极析氧(OER)和阴极析氢(HER)催化剂来降低催化反应能垒,降低制氢成本。
对于OER电催化,由于酸性条件下苛刻的反应环境,大多数非贵金属催化剂难以维持正常的运行,因而仍十分依赖Ir基和Ru基等贵金属电催化剂,为了降低成本,亟需维持高活性的同时降低贵金属载量并提高稳定性;而在碱性环境中,Ni基、Fe基等非贵金属电催化剂展现出了更有利的优势,但仍需进一步提高活性和稳定性。对于HER电催化,目前应用最广泛的是昂贵的Pt基材料,为了实现工业化应用,一方面通过调控Pt的电子结构以增强其本征活性,另一方面则需发展高性能非Pt电催化剂。近年来,已经发展了形形色色的具有新结构及高催化性能的电解水催化剂,并已成功应用于质子交换膜电解槽(PEMWE)和阴离子交换膜电解槽(AEMWE)中,展现出了强劲的工业应用前景。本文总结了山东师范大学孙旭平教授、青岛科技大学林健健教授、深圳大学蔡兴科研究员、北京大学郭少军教授、华中师范大学朱成周教授、中国科学技术大学焦淑红教授、太原理工大学桑胜波教授以及兰州大学韩新豹研究员等课题组在电解水催化剂方面的最新研究成果。
1. Nano Research:耦合双金属磷化物和氧化锰促进水解离实现高效碱性析氢电催化
过渡金属磷化物(TMPs)被认为是极具前景的碱性HER电催化剂,具有储量丰富、电子结构可调和类金属催化活性的优点。然而,由于Volmer水解离过程缓慢,活性位点与氢中间体之间的结合过强,因此造成碱性HER的催化性能仍无法满足实际需求。近年来,金属/(氢氧)氧化物在HER方面表现出了出色的性能,其可促进水解离并产生大量质子。TMPs和金属氧化物的结合可分别表现出强的M-H和M-OH结合能,从而产生强烈的相互作用来裂解水分子。作为路易斯酸物种,氧化锰在高还原电位下非常稳定,可有效分解水而不会阻碍其他活性位点的暴露。因此,将TMPs与氧化锰结合有望加速水解离动力学,从而促进碱性HER电催化的Volmer步骤。
鉴于此,电子科技大学牛晓滨教授、山东师范大学孙旭平教授和唐波教授等人通过表面改性策略在泡沫镍负载的磷化镍钴纳米阵列上修饰氧化锰纳米粒子(MnOx@NiCoP/NF),降低了催化剂的d带中心,促进了水解离和氢中间体的结合强度,实现了高效的碱性HER电催化,并在AEMWE中展现出了良好的应用前景。这项工作不仅为碱性HER电催化提供了性能优异的催化剂,而且为设计和构建用于电解水的MnOx/TMPs异质结构催化剂提供了指导。
本文要点:
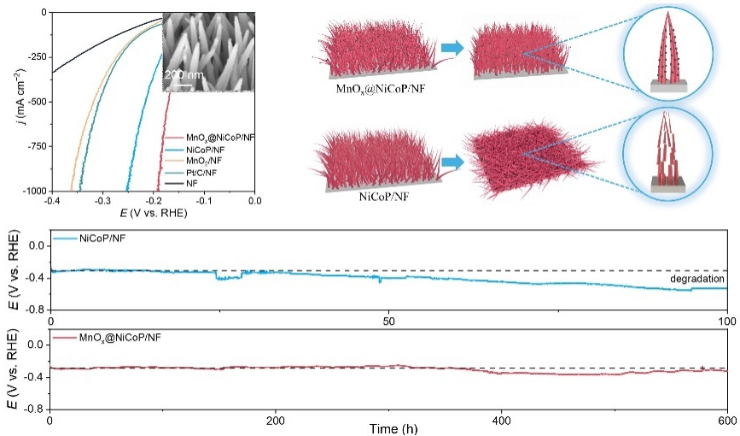
1)利用NF上NiCo层状双氢氧化物(NiCo-LDH)和MnO4-之间的自发氧化还原及随后的选择性磷化,成功实现了MnOx@NiCoP/NF的制备,其呈现出纳米颗粒修饰的纳米阵列形貌;
2)在1.0 M KOH电解质中,MnOx@NiCoP/NF在100、500和1000 mA cm-2电流密度下的过电位分别为135、171和193 mV,优于MnO2/NF和NiCoP/NF,并可在1000 mA cm-2电流密度下稳定地运行超过600小时;
3)实验和理论计算结果表明,MnOx可显著提高水解离速率,改变了金属活性位点与中间体之间的结合强度,从而有效地促进了HER过程中的本征电荷传输和H2的产生;
4)以MnOx@NiCoP/NF分别作为阴极和阳极组装AEMWE,在1.74、1.82和1.92 V的电位下就可分别实现100、200和400 mA cm-2的电流密度,并可在工业级条件下稳定地电解100小时。
Xiaolan Tang, et al., Facilitated water dissociation by coupling bimetallic phosphide with manganese oxide to enhance alkaline hydrogen evolution. Nano Res., doi: 10.26599/NR.2025.94907136.

识别二维码访问原文
2. Nano Research:室温发酵策略制备C改性非晶态NiFe合金纳米颗粒实现高效析氧电催化
在各种3d过渡金属基催化剂中,Ni和Fe在碱性电解质中显示出巨大的OER催化潜力。NiFe合金的协同效应可进一步调节局部活性位点的电子结构,从而改善OER动力学。然而,NiFe合金在高浓度碱性电解质中容易遭到腐蚀,并在高电位下难以稳定。此外,在电化学测试过程中还会造成NiFe合金纳米颗粒的聚集,减少活性位点的数量并阻碍传质。因此,亟需开发一种有效的方法来优化NiFe合金的OER活性和稳定性。与晶态合金相比,非晶合金表现出更多的缺陷和空位,导致作为电化学反应活性位点的悬空键浓度更高。使用C作为催化剂改性策略可以进一步调节催化剂的尺寸并暴露更多活性位点,有望促进电子转移并增强催化活性。此外,C的掺杂还有助于稳定晶体结构并提高稳定性。
鉴于此,青岛科技大学林健健教授等人采用室温发酵策略制备了C修饰NiFe非晶合金催化剂(C-NiFe(BP)),显著提升了碱性OER催化活性和耐久性。这项工作为合成稳定的C改性NiFe非晶合金催化剂提供了一种创新方法,并进一步促进了高性能OER催化剂的发展。
本文要点:
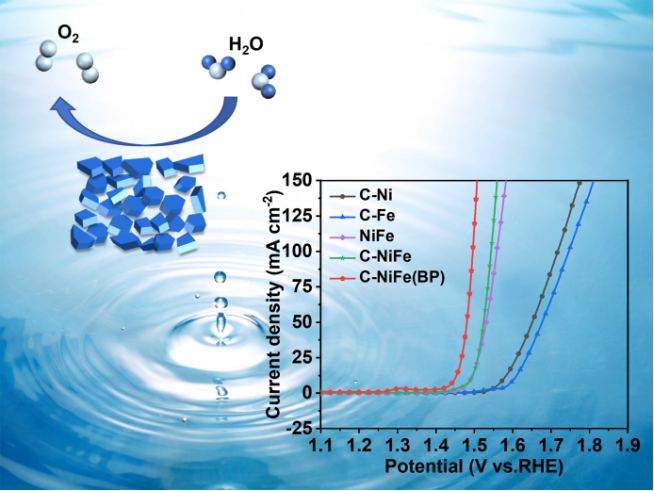
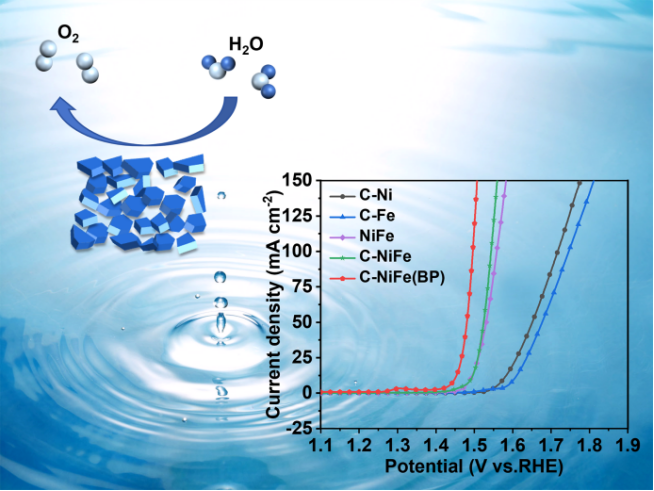
1)在室温发酵的条件下,以葡萄糖作为碳源,KBr作为模板剂,以相对低的温度下直接一步热处理即可获得C-NiFe(BP),室温发酵过程使材料变得更蓬松,从而增大了比表面积;
2)由于碳的改性,催化剂的层间距被显著增大,从而使其暴露出更多的活性位点,促进了元素之间的电荷转移,提高了OER活性;
3)在1.0 M KOH电解质中,C-NiFe(BP)在10 mA cm-2电流密度下的OER过电位仅为219.7 mV,Tafel斜率为43.17 mV dec-1,超越了大多数已报道的NiFe催化剂,并在循环1000圈或20小时以后,活性几乎没有衰减;
4)利用葡萄糖作为碳源有助于提高催化剂的稳定性,而葡萄糖的发酵可进一步增加活性位点的数量,有助于提升OER性能。
Yanzhen Qiu, et al., C-modified amorphous NiFe alloy nanoparticle via simple room temperature fermentation for efficient oxygen evolution reaction.Nano Res., doi: 10.26599/NR.2025.94907019.

识别二维码访问原文
3. Nature Nanotechnology:Ir单原子与有机分子和钴铁氢氧化物的面外配位增强析氧电催化
Co、Fe和Ni基金属氢氧化物因其成本效益和高的催化活性成为了极具前景的OER候选催化剂。为了进一步降低OER过电位,近年来已经提出了形貌调控、缺陷设计、异质界面构筑、元素掺杂和单原子负载等策略。其中,将贵金属单原子负载到金属羟基氧化物/氢氧化物上已被证明可以有效地将OER过电位降低到200 mV以下。但需要进一步的研究来充分利用单原子配位、成键和催化性能之间的相互作用。目前,已经通过单原子催化剂的面外配体配位(轴向配位)实现了各种电化学反应电催化活性的提升,因此,将该策略应用于金属羟基氧化物/氢氧化物上的贵金属单原子有望显著提高OER性能,但仍缺乏相关研究。
鉴于此,深圳大学蔡兴科研究员和德国拜罗伊特大学Francesco Ciucci教授等人在CoFe氢氧化物纳米片上制备了与二甲基咪唑(MI)面外配位的Ir单原子(Ir1/(Co,Fe)-OH/MI),实现了OER电催化性能的显著提升。这项工作的制备方法可以扩展到其他贵金属,如Pt、Pd和Ru,为研究各种催化反应的催化机制提供了一个新的思路。
本文要点:
1)首先将含有Co2+和MI分子的水溶液在泡沫镍上生长出花瓣状Co-MI片材,随后将这些片材浸入含有Co2+、Fe3+和Ir3+的乙二醇/水溶液中,即可实现Ir1/(Co,Fe)-OH/MI的制备,该材料具有纳米片结构,并且存在孤立分散的Ir原子,负载量为0.33 wt.%;
2)结构表征结果表明,由于Ir与MI的部分配位,导致Ir1/(Co,Fe)-OH/MI中Ir的价态位于0和+4之间,且比Ir1/(Co,Fe)-OH的高,同时MI配体与Ir单原子呈现出面外配位的特点;
3)在1.0 M KOH电解质中,Ir1/(Co,Fe)-OH/MI在10 mA cm-2电流密度下的OER过电位低至179 mV,法拉第效率超过98%,其面积比活性和质量活性分别超过商业IrO2催化剂16.4和58.4倍,并在不同电流密度下均可稳定运行,且具有连续120小时的稳定性;
4)理论计算结果表明,与MI的面外配位导致Ir(N)-CoFe中Ir和Co位点的电子重排,使d带中心上移,提高了Ir和Co的氧化态,从而增强了OER中间体的吸附,提升了催化性能;
5)在AEMWE中,不对称Ir1/(Co,Fe)-OH/MI||20% Pt/C电解槽在低于500 mA cm-2时表现出优异的性能,而对称Ir1/(Co,Fe)-OH/MI||Ir1/(Co,Fe)-OH/MI电解槽在高于500 mA cm-2时性能更佳,两种电解槽均可在500 mA cm-2下稳定运行超过150小时;
6)MI 面外配位的策略对于贵金属单原子的制备具有高度普适性,通过简单的两步法,可实现Pt1/(Co,Fe)-OH/MI、Pd1/(Co,Fe)-OH/MI和Ru1/(Co,Fe)-OH/MI等的制备,均表现出单原子分散在多孔氢氧化物上的特点。
Jie Zhao, et al., Out-of-plane coordination of iridium single atoms with organic molecules and cobalt-iron hydroxides to boost oxygen evolution reaction. Nat. Nanotech., doi: 10.1038/s41565-024-01807-x.

识别二维码访问原文
4. Nature Communications:晶格Pb的钉扎效应抑制Pb-RuO2晶格氧反应性实现超稳定工业级电解水
由于缺乏高效、低成本的酸性OER催化剂,PEMWE的大规模应用仍受到严重限制,最大的瓶颈在于如何在高电流密度下增强催化剂在强酸性条件下的耐久性。到目前为止,稳定性强、活性高的Ir基材料被认为是最佳的酸性OER催化剂,然而,商用PEMWE中Ir的使用量通常很高,这限制了它们的大规模应用。最近,Ru基催化剂因其更高的OER活性和更低的成本而备受关注,然而,即使在低电流密度下运行,Ru基催化剂也总是表现出较差的稳定性,这远低于PEMWE对高电流密度的要求(>500mA cm-2)。主要原因可归因于Ru基催化剂的高Ru-O共价性和不可避免的晶格氧机制(LOM),这增加了晶格氧在反应过程中的参与,从而导致Ru位点的过度氧化和催化剂不可抑制的结构坍塌。因此,开发一种有效的策略来降低晶格氧的反应性对于提高Ru基催化剂的耐久性至关重要,但仍面临巨大挑战。
鉴于此,北京大学郭少军教授和骆明川研究员等人采用大尺寸和耐酸的晶格铅(Pb)作为第二元素来诱导钉扎效应,有效地缩短了氧原子的移动通道,从而降低了Ru氧化物中晶格氧的反应性,在酸性电解质中展现出超稳定的OER性能,并可用于PEMWE中。这项工作有助于促进RuO2在PEMWE中的实际应用。
本文要点:
1)通过一锅葡萄糖吹扫法实现了Pb-RuO2催化剂的制备,其呈现出金红石型结构,由于大尺寸Pb的引入,晶格发生了膨胀,且有助于打破RuO2的对称性,减弱了Ru-O键的共价性;
2)在0.5 M H2SO4电解质中,Pb-RuO2在10和100 mA cm-2电流密度下的过电位分别为 188 ± 2 和255 ± 4 mV,优于采用类似方法制备的纯RuO2和商业RuO2,经过1100小时的长期稳定性测试以后,电位的衰减率仅为19 μV h-1;
3)XAFS、原位微分电化学质谱和理论计算结果表明,由于6s-2p-4d轨道杂化导致的Ru-O共价性降低,Pb的参与会削弱晶格氧的反应性,从而抑制Ru的过度氧化,保持良好的晶体结构,因而提高了催化剂的稳定性;
4)以Pb-RuO2作为阳极、商业Pt/C作为阴极组装PEMWE,在1.582、1.688和1.848 V的电位下就可实现500、1000和2000 mA cm-2的电流密度,并可在500 mA cm-2电流密度下稳定地运行超过250小时,电位的衰减速率仅为17 μV h-1,即使在高达1000 mA cm-2电流密度下,电位的衰减速率也仅87 μV h-1。
Chenhui Zhou, et al., Pinning effect of lattice Pb suppressing lattice oxygen reactivity of Pb-RuO2 enables stable industrial-level electrolysis. Nat. Commun., doi: 10.1038/s41467-024-53905-y.

识别二维码访问原文
5. Nature Communications:具有晶格氢参与的钯氢化物金属烯气凝胶实现高效析氢电催化
由于类Pt的电子结构和高储量,Pd基催化剂作为Pt的替代品近年来在HER电催化中引起了极大的关注。然而,Pd和H之间的强结合不利于H2的解吸,导致HER性能较差。深入了解催化剂的HER催化机制对于提高催化效率具有重要意义。酸性HER途径通常涉及Volmer步骤,该步骤通过电化学还原产生吸附氢中间体(H*),然后是电荷转移Heyrovsky过程或Tafel重组过程产生H2。基于Sabatier原理,理想的HER电催化剂应具有接近零的氢吸附自由能(∆GH*)。氢结合能太强或太弱都会导致活性位点中毒或质子补充效率低下,大大降低HER的反应动力学。此外,单相催化剂中的氢吸附和解吸通常发生在单个活性位点,难以克服Sabatier原理的局限性。因此,开发具有多个催化位点的催化剂有望突破现有HER催化机制的瓶颈。
鉴于此,华中师范大学朱成周教授等人提出了β-Pd氢化物金属烯(β-PdHene)气凝胶的晶格氢参与机制,从而分离吸附和解吸位点,实现了HER性能的显著增强。这项工作可以避开传统机制下的催化剂设计原则,为合理开发高性能HER催化剂开辟了一条新途径。
本文要点:
1)首先利用CO诱导凝胶化策略制备Pd金属烯(Pdene)凝胶,然后通过DMF分解原位形成的氢原子插入Pdene凝胶,获得β-PdHene凝胶,最后经纯化和超临界CO2干燥,即可获得单片β-PdHene气凝胶;
2)所制备的β-PdHene气凝胶具有分层多孔结构,厚度约为1.26 nm,相当于5-6个原子层,并表现出良好的存储稳定性;
3)在0.5 M H2SO4电解质中,β-PdHene气凝胶在10和50 mA cm-2电流密度下的HER过电位分别为20和66 mV,低于Pdene和商业Pt/C,同时展现出良好的稳定性;
4)电化学测试结果表明,β-PdHene气凝胶在催化HER过程中,可以实现更慢的氢吸附动力学,在速率决定步骤中可实现氢或质子的转移;
5)原位微分电化学质谱和理论计算结果证实了晶格氢在HER过程中的参与过程,相邻的H*会发生迁移并与表面活化的晶格氢结合形成H2,从而促进H2的解吸。
Hengjia Wang, et al., Pd hydride metallene aerogels with lattice hydrogen participation for efficient hydrogen evolution reaction. Nat. Commun., doi: 10.1038/s41467-024-54601-7.

识别二维码访问原文
6. Journal of the American Chemical Society:核/壳异质结构的超薄保形消耗层实现高效稳定酸性水氧化
Ru基催化剂在酸性电解质和强氧化环境下的快速降解限制了其在PEMWE中的实际应用。在OER的高电位条件下,Ru位点很容易被过度氧化,形成高价可溶性Ru物种(如RuO4),导致Ru基催化剂的溶解。尽管已经提出了包括元素掺杂、应变工程和结构调控等的各种策略来抑制在OER过程中Ru物种的过度氧化,但迄今为止报道的大多数Ru基催化剂仍难以权衡酸性OER过程的活性和稳定性。异质结构可利用费米能级排列驱动电荷转移,成为了提高催化剂电催化稳定性和活性的极具前景的策略。然而,仍存在基底材料易腐蚀等问题,这通常会导致异质结构的破坏,降低催化活性。此外,异质结构催化剂内的大多数活性位点也通常被包裹在材料内部,无法与反应物分子直接接触。因此,设计一种用于高效稳定的酸性OER异质结构催化剂仍面临巨大挑战。
鉴于此,中国科学技术大学焦淑红教授、曹瑞国教授、江俊教授和宁波东方理工大学M. Danny Gu教授等人提出了一种超薄的保形消耗层,其可附着在核/壳RuCo/RuCoOx肖特基异质结上,不仅可最大限度地提高活性位点与反应物分子的接触,而且大幅提高了酸性OER的耐久性和本征活性。这项工作提出了异质结构催化剂的合理设计,有助于促进异质结构催化剂在电化学应用中的广泛发展。
本文要点:
1)为了构建具有暴露消耗层的异质结,设计了一种可控的热处理氧化方法,可以通过精确调节RuCo合金纳米球的氧化条件来制备RuCo/RuCoOx核/壳异质结构;
2)不同温度热处理的RuCo/RuCoOx核/壳异质结构均展现出纳米球形态,直径约为40 nm,其中在250°C处理的RuCo合金被厚度约为1.5 nm的保形壳层包围,从而形成封闭的核/壳异质结构;
3)在0.5 M H2SO4电解质中,RuCo/RuCoOx在10 mA cm-2电流密度下的OER过电位仅170 mV,在连续运行2500小时后,活性保持率高达94.4%;
4)以RuCo/RuCoOx为阳极,商业Pt/C作为阴极组装PEMWE,仅需要1.784 V的电位就可实现1 A cm-2的电流密度,并可在200 mA cm-2电流密度下稳定地运行超过200小时;
5)工况条件下的同步辐射和理论计算结果表明,肖特基异质结引起的晶格应变和电荷转移调控了活性位点的电子结构,从而改变了OER途径并抑制了Ru物种的过度氧化,此外,RuCo/RuCoOx封闭的核/壳结构还确保了肖特基异质结在酸性OER条件下的结构完整性。
Yang Liu, et al., Ultrathin and conformal depletion layer of core/shell heterojunction enables efficient and stable acidic water oxidation.J. Am. Chem. Soc., doi: 10.1021/jacs.4c07995.
识别二维码访问原文

7. Advanced Materials:局部无序诱导PtZn金属间化合物的超快氢溢流实现高效析氢电催化
由于几何和电子效应的协同作用,原子有序的金属间化合物往往比无序固溶体合金表现出更高的结构稳定性和活性。Zn具有较低的熔点和沸点,很容易通过退火形成有序的金属间PtZn。然而,Zn的高氢过电位导致PtZn过高的H*中间体吸附吉布斯自由能(ΔGH*),从而影响HER催化活性。已经通过非金属元素掺杂、晶格应变工程和三金属有序金属间化合物的设计来解决PtZn的活性问题,但仍无法从本质上克服Zn的H排斥特性。原子尺度的氢溢流效应已被用于通过增强单相催化剂中的质子传输来提高HER活性。众所周知,Pt位于酸性HER火山图的顶部,而Zn排斥H。当Pt和Zn原子同时暴露在同一层时,可能会发生原子尺度的氢溢流,涉及氢在Pt位点的吸附和随后从相邻的Zn位点的解吸。然而,关于如何调控Pt和Zn原子的原子排列以提高PtZn的催化活性的研究鲜有报道。
鉴于此,太原理工大学桑胜波教授、王美玲副教授、北京理工大学周家东教授和南京晓庄学院胡应杰博士等人利用Zn在高温下的挥发性,提出了一种限域高温热解策略,以诱导有序金属间PtZn(I-PtZn)的局部无序化,实现了超短的氢溢流通道,显著提高了HER电催化性能。这项工作为有序金属间PtZn中局部Zn无序的设计以实现高效电催化提供了新的思路。
本文要点:
1)首先通过ZIF-8的热解制备氮掺杂碳负载的Zn原子(Zn1@NPC),随后Zn原子在高温下的挥发有助于捕获游离[PtCl6]2-离子,促进Pt原子的锚定(Zn1Pt1@NPC),N-Zn键强的减弱进一步促进了PtZn金属间化合物的形成,在该过程中,Zn蒸汽有利于形成被Zn隔离的连续Pt原子,同时Pt原子可能会随着Zn的逃逸而渗透到Zn空位中,导致PtZn的部分无序化;
2)在0.5 M H2SO4电解质中,I-PtZn/NPC在10 mA cm-2电流密度下的HER过电位仅2.3 mV,远低于PtZn/NPC和商业Pt/C,将其作为阴极,商业IrO2作为阳极组装PEMWE,仅需1.60 V的电位就可实现1 A cm-2的电流密度,并可稳定地运行100小时;
3)原位实验和理论计算结果表明,由于Pt原子对Zn原子的局部替代,I-PtZn表现出有利的氢溢流效应,具体而言,Pt充当质子富集位点,热中性Pt-Zn桥位点充当从Pt位点到相邻Zn位点的H迁移介质,Zn位点有利于最终的H2解吸;
4)通过超短H溢流路径(Pt位点→Pt-Zn桥位点→Zn位点),I-PtZn@NPC在酸性电解质中表现出了优异的HER活性和耐久性。
Meiling Wang, et al., Ultrafast H-spillover in intermetallic PtZn induced by the local disorder for excellent electrocatalytic hydrogen evolution performance. Adv. Mater., doi: 10.1002/adma.202409575.

识别二维码访问原文
8. ACS Nano:亚纳米Pt-W双金属团簇实现高效碱性析氢电催化
将两种不同的金属组合成超小的双金属纳米粒子(UBN)时,两种金属之间的协同效应和量子尺寸效应有助于优化特定的反应途径并优化催化性能。亚纳米双金属簇(SBC)的尺寸比UBN更小,不仅可充分利用双金属协同效应,而且可实现近100%的原子利用效率,从而表现出比单原子催化剂和UBN更高的催化活性。近年来,已经发展了多种合成SBC的方法,但所得材料通常存在传质差或活性位点暴露不足的问题。此外,SBC的种类也有限,不仅限制了催化性能,而且限制了广泛的应用。因此,制备具有窄尺寸分布的SBC,并且在载体上充分暴露活性位点仍面临巨大挑战。
鉴于此,兰州大学韩新豹研究员、彭勇教授和云南大学张宏副教授等人发展了一种普适性的“闭孔金属有机框架材料表面化学限域”策略,通过使用更廉价的无孔且带正电的金属有机框架(MOF)与带负电的多金属氧酸盐(POM),利用静电作用形成MOF表面化学限域POM,经热解形成了氮掺杂碳(NC)载体表面充分暴露的SBC催化剂,在碱性电解质中实现了卓越的HER催化性能。这项工作所报道的策略可扩展到其他MOF和没有孔或有限孔隙率的配位结构,从而设计和制备金属物种完全暴露的功能材料,有望在各种领域表现出良好的应用前景。
本文要点:
1)利用无孔且带正电的[ZnATZ]2+ MOF作为载体与带负电的[H3PtW6O24]5- POM相结合,通过热解制备了PtW/NC催化剂,[H3PtW6O24]5- POM与闭孔MOF [ZnATZ]2+之间的静电作用可避开两个常见的障碍:(i) 防止金属物种在NC表面聚集成许多大的纳米颗粒,(ii) [ZnATZ]2+的闭孔性质在退火过程中成功地阻止了POM及其衍生金属物种进入孔隙,在内部形成大的团簇或纳米颗粒;
2)在所制备的催化剂中,球状NC表面负载有高度分散,且平均颗粒大小在0.81 nm左右的PtW SBCs;
3)在1.0 M KOH电解质中,PtW/NC在10 mA cm-2电流密度下的HER过电位仅4 mV,质量活性高达19.7 A mg-1,在连续运行140小时以后,活性几乎没有衰减;
4)理论计算结果表明,W的引入降低了PtW团簇上质子的吸附强度,增强了H2O和OH*的吸附,从而在PtW/NC上实现了优异的碱性HER电催化性能。
Shoushun Chen, et al., Subnanometric Pt-W bimetallic clusters for efficient alkaline hydrogen evolution electrocatalysis.ACS Nano, doi: 10.1021/acsnano.4c13743.

识别二维码访问原文

https://blog.sciencenet.cn/blog-3563286-1464101.html
上一篇:喜讯|《电力能源汇刊(英文)》被ESCI数据库收录
下一篇:Nano Res.[合成]│袁小明课题组:与WS₂耦合的间隙可调等离子体纳米腔的激子调控和载流子动力学