博文
配位引起的轴手性双核 Pt(II) 配合物的外消旋化机制
|
Density Functional Theory Calculation on the Racemization Mechanism of Metal-Induced Axial Chirality: Axial Rotation vs Coordination Geometry Change
Wenjing Dong, Bao Zhang, Bo Tu, Qingping Yang, Xiangge Zhou, Haifeng Xiang*
Inorg. Chem. 2026 doi: 10.1021/acs.inorgchem.5c05863
Abstract
Axially chiral molecules exhibit atropisomerism, with racemization energy barriers typically governed by steric hindrance. This study investigates the racemization mechanism of metal-induced axial chirality in four-coordinate, square-planar binuclear Pt(II) complexes bearing one cyclometalated sym-tetraacetylethane bridging ligand and two cyclometalated 2-phenylpyridine ligands. Notably, the experimental racemization barrier (34.4 kcal/mol) of the complex featuring four bulky methyl groups at the ortho-positions of its chiral axis is lower than that of the classical axial chiral reference, 1,1′-binaphthol (40.5 kcal/mol), suggesting an alternative racemization pathway beyond a simple axial rotation. Given the relatively low bond dissociation energies of coordination bonds, we employed density functional theory to simulate a novel pathway involving a change in coordination geometry from four- to three-coordinate. The resulting three-coordinate intermediate, which contains a monodentate bridging ligand, can readily undergo single-bond rotation to form its enantiomer. The calculated energy barriers for this process range from 34.0 to 40.4 kcal/mol using various functionals and basis sets. Although these values are somewhat higher than most reported racemization barriers of chiral-at-metal complexes (<32 kcal/mol), they substantiate the feasibility of the proposed mechanism. Consequently, this study offers valuable insights for the rational design of chiral-at-metal complexes.
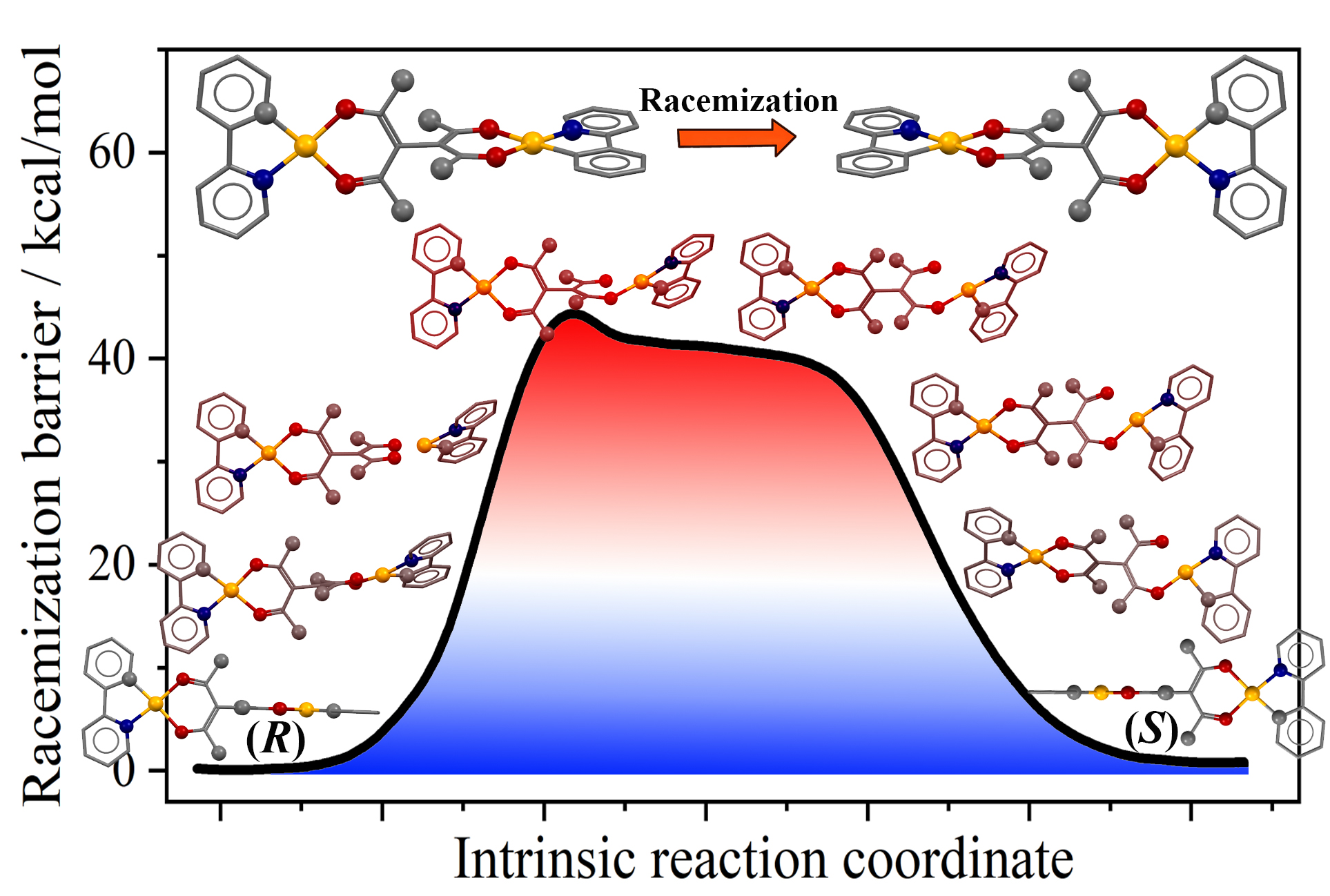
这篇发表于 Inorganic Chemistry 的文章《Density Functional Theory Calculation on the Racemization Mechanism of Metal-Induced Axial Chirality: Axial Rotation vs Coordination Geometry Change》由 Wenjing Dong 等人完成,通讯作者为 Haifeng Xiang(四川大学)。研究聚焦于金属诱导轴手性双核 Pt(II) 配合物的外消旋化机制,并通过密度泛函理论(DFT)揭示了与经典有机轴手性分子(如 BINOL)完全不同的外消旋路径。
以下是对该文的深度解读与分析:
一、研究背景与核心问题1. 轴手性与外消旋
轴手性分子因围绕单键的旋转受阻而产生阻旋异构现象。
传统有机轴手性分子(如联萘酚 BINOL)的外消旋能垒(ΔG)主要由空间位阻决定。
一般要求 ΔG > 22.2 kcal/mol 可拆分,> 29.4 kcal/mol 可稳定存在。
2. 实验中的反常现象
作者之前合成的双核 Pt(II) 配合物 Pt2 具有四个邻位甲基,空间位阻极大。
按理其外消旋能垒应高于 BINOL(40.5 kcal/mol),但实验测得 ΔG_exp = 34.4 kcal/mol,反而更低。
提示可能存在非轴向旋转的外消旋机制。
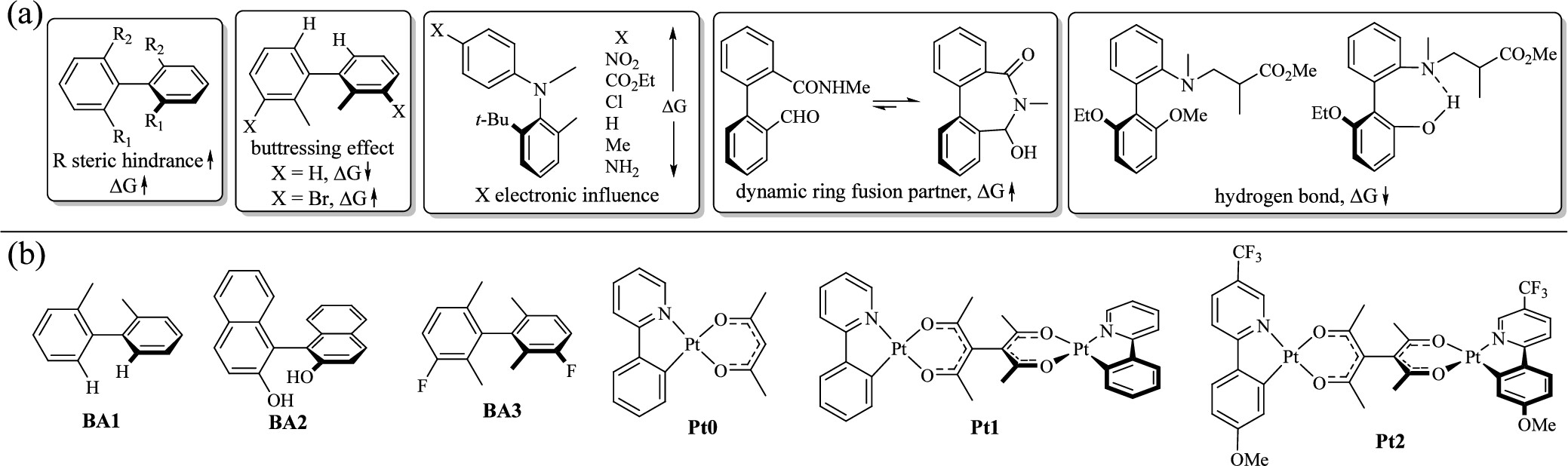
二、研究目标与创新点目标:
探究 Pt2 及其模型化合物 Pt1 的外消旋机制,解释其能垒低于预期的问题。
创新点:
提出并验证了一种通过配位几何变化(四配位→三配位)实现外消旋的新机制,而非传统的轴向旋转。
三、计算方法与策略软件与泛函:
使用 Gaussian 09/16。
主要泛函:PBE1PBE、B3LYP、WB97XD、TPSSH、M06、M062X。
基组:C、H、O、N、F 用 6-31G*、6-311G**、def2SVP、def2TZVP;Pt 用 SDD 赝势。
溶剂模型:PCM(o-二甲苯)。
色散校正:GD3、GD3BJ。
分析方法:IRC、频率分析、过渡态搜索、RMSD 结构对比。
四、关键发现与机制解析1. 有机分子 BA1–BA3 的对照研究
BA1、BA2 的外消旋通过轴向旋转实现,ΔG_cal 与实验值吻合良好。
BA3 由于四个甲基的空间位阻过于均衡,导致旋转受阻,能垒极高(123.1 kcal/mol),可能发生 C–C 键断裂。
Pt1 的松弛扫描与 BA3 相似,提示其可能也不通过轴向旋转外消旋。
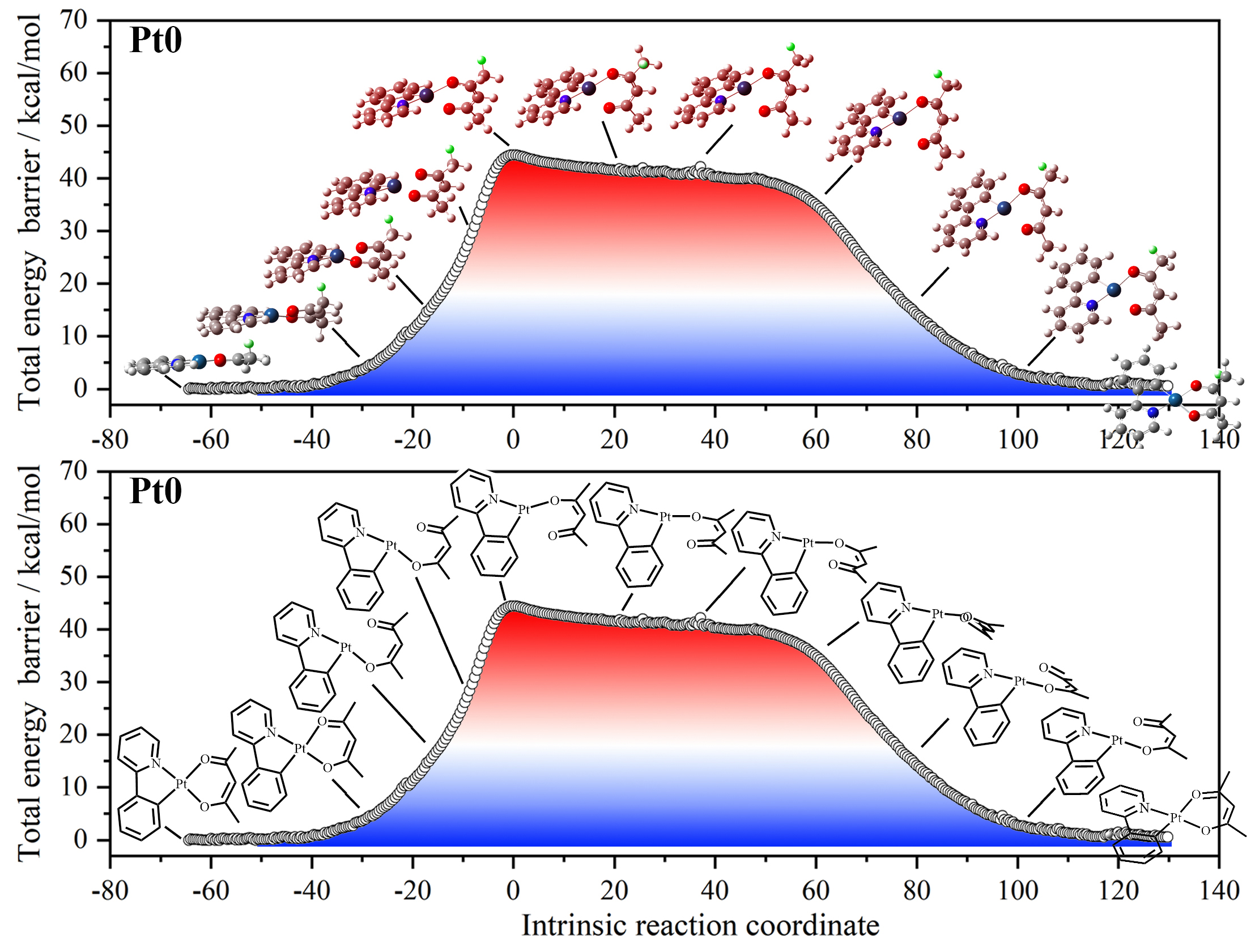
2. Pt0 模型体系验证几何变化机制
Pt0 为单核模型配合物,结构简单。
在过渡态中,一个 Pt–O 键从 2.115 Å 拉长至 2.646 Å,形成三配位三角几何。
IRC 显示 O^O 配体可绕 Pt 旋转 180°,实现构型翻转。
ΔG_cal = 41.8 kcal/mol,与 Pt–O 键断裂能一致。
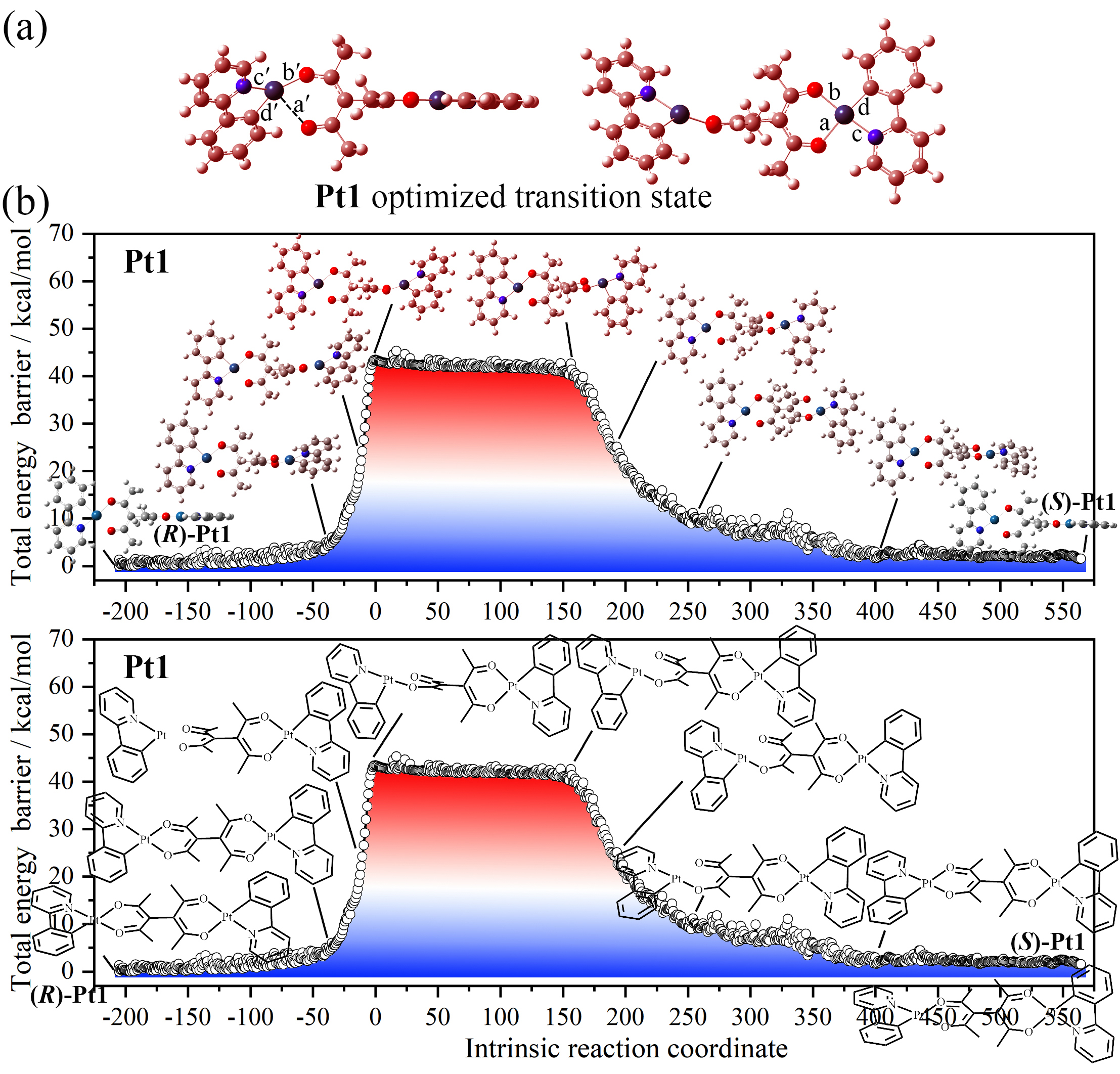
3. Pt1 与 Pt2 的过渡态与 IRC 分析
过渡态中,一个 Pt 中心保持四配位,另一个变为三配位(Pt–O 键断裂至 >2.6 Å)。
IRC 确认该过渡态连接 (R)- 和 (S)- 构型,是正确的外消旋路径。
Pt1 ΔG_cal = 40.0 kcal/mol,Pt2 ΔG_cal = 40.9 kcal/mol(气相)。
4. 溶剂效应与泛函依赖性
在 PCM 溶剂模型下,ΔG_cal 降低至 34.0–40.4 kcal/mol,与实验值 34.4 kcal/mol 高度吻合。
M06、M062X 泛函表现最佳,尤其结合 GD3 色散校正。
溶剂效应主要来自对过渡态(极性更大)的稳定作用,而非配位。
五、结论与意义结论:
Pt1 和 Pt2 的外消旋并非通过轴向旋转,而是通过配位几何变化实现:
四配位 → 三配位(Pt–O 键断裂)
单齿桥联配体自由旋转 → 构型翻转
再配位恢复四配位结构
计算能垒与实验值一致,验证了该机制的合理性。
意义:
首次系统研究了平面四方双核 Pt(II) 配合物的几何变化外消旋机制。
提出了一种新的金属诱导轴手性外消旋路径,为手性金属配合物的设计提供了新思路。
强调在金属配合物中,配位键的弱键特性可诱导出不同于有机分子的立体化学行为。
六、值得关注的点
机制普适性:该机制是否适用于其他 d⁸ 金属(如 Pd、Ni)或其他配体类型?
实验验证手段:是否可通过变温 NMR、动力学同位素效应等手段进一步验证?
应用前景:可用于设计构型稳定但可控外消旋的手性金属配合物,应用于不对称催化、CPL 材料等领域。
https://blog.sciencenet.cn/blog-581887-1526595.html
上一篇:构建半灯笼状双核Pd(II)配合物:金属诱导平面手性、手性自分类及近100%磷光量子产率
下一篇:四配位平面四边形配合物的配位构型热稳定性探究
