博文
缺氧和肺循环中离子通道的氧化还原调控
||
缺氧和肺循环中离子通道的氧化还原调控
缺氧条件下肺循环中离子通道的氧化还原调控
意义:肺部急性缺氧会引发缺氧性肺血管收缩,从而优化气体交换。与之相反,慢性缺氧会触发病理性血管重构,进而导致肺动脉高压;缺血则可造成血管损伤,最终引发肺水肿。
最新研究进展:细胞氧化还原状态对离子通道表达与门控的调控是公认机制;然而,缺氧条件下活性氧(ROS)水平究竟升高还是降低,目前仍存在争议。部分离子通道(如电压门控钾通道、瞬时受体电位通道6)的氧化还原调控已有详细报道,但整体而言,离子通道氧化还原调控的相关信息仍较为匮乏。
关键问题与未来方向:除缺氧时ROS生成增多与减少的争议外,本文旨在阐述并解析为何不同氧化剂在不同条件下既可激活也可抑制通道活性。尽管影响通道门控的上游通路已有较多阐释,但我们仍需更深入地理解氧化还原蛋白修饰,才能阐明离子通道氧化还原调控的复杂机制。在此背景下,本文总结了缺氧诱导ROS介导的肺循环离子通道信号通路的现有研究成果。
Veit F, Pak O, Brandes RP, Weissmann N. Hypoxia-dependent reactive oxygen species signaling in the pulmonary circulation: focus on ion channels. Antioxid Redox Signal. 2015 Feb 20;22(6):537-52.
引言
肺泡缺氧对肺循环的影响可分为三个阶段:
(1)急性期(30秒~<20分钟);
(2)持续期(>30分钟至数小时~数天):此阶段通过缺氧性肺血管收缩(HPV),在局部肺泡缺氧时使血流灌注与肺泡通气相匹配;
(3)慢性期:以顽固性血管收缩与血管重构(中膜肥厚)为特征,最终诱发肺动脉高压(PH)(图1)。
肺泡上皮细胞通常处于100 mmHg氧分压环境,氧分压极少低于40 mmHg。在弥漫性肺泡缺氧时,急性与持续期反应同样参与肺动脉高压的发生发展。大量证据表明,这三个阶段至少部分由不同机制调控。尽管内皮细胞可通过释放血管活性物质调节血管收缩,但该反应是肺动脉平滑肌细胞(PASMC)作为效应细胞所特有的。
已知氧水平变化会伴随肺动脉平滑肌细胞内ROS生成改变,ROS被认为是缺氧依赖性信号的关键介质。尽管氧感受机制尚不明确,多项证据表明线粒体与NADPH氧化酶是主要的ROS来源。在哺乳动物细胞多种可生成ROS的系统中,线粒体呼吸链与NADPH氧化酶参与调控急性缺氧时的缺氧性肺血管收缩,以及慢性缺氧时的细胞增殖与中膜肥厚。
尽管氧化还原信号被认为在缺氧性肺血管收缩与血管重构中至关重要,但目前仍存在争议:ROS水平升高或降低究竟哪一种触发了上述过程。ROS可与DNA、脂质及多肽发生反应,但其特异性仅体现在对蛋白的作用上。高浓度ROS可引发多种氨基酸修饰(巯基氧化、精氨酸与赖氨酸残基氧化、甲硫氨酸氧化)、分子交联及蛋白在多分子复合物中的捕获。与破坏蛋白功能的不可逆修饰不同,生理水平的ROS可通过与氨基酸特异性结合调控蛋白功能。
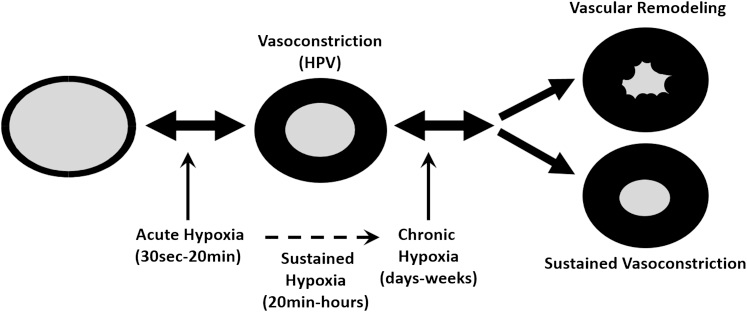
图1肺泡缺氧对肺循环影响的示意图。肺泡缺氧对肺循环的影响分为三阶段:
(1)急性期(30秒~20分钟);
(2)持续期(20分钟~数小时);
(3)慢性期(数天~数周)。
急性缺氧数秒内即可引发缺氧性肺血管收缩,使血流灌注与肺泡通气匹配。在弥漫性持续与慢性肺泡缺氧时,血管收缩通过中膜肥厚(血管重构)被形态学固定,进而诱发肺动脉高压。
肺血管缺氧反应中离子通道的参与,揭示了L型钙通道与钾通道是重要调控分子。后续发现的多种其他钾、钙通道同样参与缺氧性肺血管收缩与慢性缺氧诱导的肺动脉高压。这些离子通道对氧化还原变化高度敏感。目前存在两种对立模型:
第一种模型认为,线粒体ROS释放减少介导电压门控钾通道关闭,进而激活血管收缩、促增殖与抗凋亡信号级联。该假说基于缺氧时肺循环ROS生成增多或减少的相反观察结果。ROS的潜在来源(线粒体或NADPH氧化酶)及其下游靶点同样存在争议(图2)。
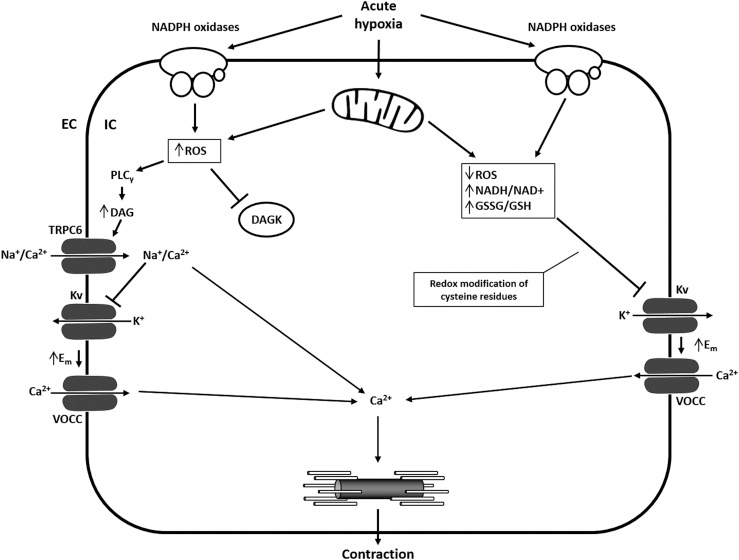
图2急性缺氧对ROS生成与电压门控钾通道调控的对立模型。
关于急性缺氧对肺动脉平滑肌细胞去极化的影响,存在两种模型:
右侧模型:ROS(主要来自线粒体)释放减少介导电压门控钾通道关闭,激活血管收缩。
第二种模型:线粒体和/或NADPH氧化酶产生的ROS增多,通过二酰甘油(DAG)介导激活瞬时受体电位通道6,引发Na⁺与Ca²⁺内流,Na⁺进而抑制电压门控钾通道。
DAG:二酰甘油;DAGK:二酰甘油激酶;EC:细胞外;Em:膜电位;IC:细胞内;Kv:电压门控钾通道;PASMC:肺动脉平滑肌细胞;ROS:活性氧;TRPC6:瞬时受体电位通道6;VOCC:电压门控钙通道。
第二种模型认为,线粒体或NADPH氧化酶产生的ROS增多,通过多种膜通道(包括电压门控钾通道、瞬时受体电位通道、L型钙通道)和/或细胞内钙释放触发上述反应(图2)。两种模型的差异可由亚细胞区室基础氧化状态不同的研究解释:急性缺氧时,胞质与膜间隙ROS生成增加,而线粒体基质ROS生成减少。
常氧条件下,线粒体复合体Ⅰ与Ⅲ产生基础水平超氧阴离子(与肺泡氧分压正相关)。复合体Ⅰ与复合体Ⅲ Q₁侧产生的超氧阴离子进入基质,复合体Ⅲ Q₀侧产生的超氧阴离子进入膜间隙,随后转化为过氧化氢(H₂O₂),可穿过线粒体外膜进入胞质。
除线粒体呼吸链外,常氧时NADPH氧化酶家族局部产生的ROS可启动生理生长因子信号所需的多种细胞反应。然而,关于急、慢性缺氧时ROS生成增多或减少,均有相关报道。
离子通道介导缺氧时肺血管张力的调控已被广泛认可。无论ROS升高或降低,L型通道介导的Ca²⁺内流与肌浆网钙释放均会导致胞内Ca²⁺升高,启动并维持肺血管平滑肌细胞(VSMC)的缺氧性收缩。
基于此,本文综述将聚焦急、慢性缺氧时离子通道的氧化还原调控作用。受限于篇幅,本文无法涵盖该领域所有研究;在肺特异性文献不足时,部分内容也会参考非肺部研究。
钾通道
钾通道介导K⁺跨细胞膜转运,对动作电位模式、上皮功能、细胞容积调控、激素分泌及细胞膜电位调节至关重要。K⁺顺电化学梯度传导,通道对K⁺具有高度选择性。所有已知钾通道均具有高度选择性的孔道结构,可分为五个亚类,主要区别在于门控激活方式:
1. 内向整流钾通道(Kir):包括经典内向整流、G蛋白门控、ATP敏感型(KATP)及K⁺转运通道;
2. 双孔道钾通道(K2P):受pH、温度、脂肪酸、电压与膜牵张调控;
3. 电压门控钾通道(Kv);
4. Slo家族(KCa):电导极大,含大电导钙激活钾通道(BK);
5. 小电导钙激活钾通道家族(SKCa)。
肺血管与肺动脉平滑肌中已鉴定出四种主要钾通道:电压门控钾通道、钙激活钾通道、内向整流钾通道、双孔道钾通道。在肺动脉平滑肌细胞中,钾通道激活后K⁺外流导致膜超极化,进而引发血管舒张;反之,钾通道抑制导致去极化与血管收缩。
钾通道抑制与K⁺内流(实质为K⁺电导降低)可启动膜去极化、电压门控钙通道激活与血管收缩。该作用在肺动脉平滑肌细胞中主要由电压门控钾通道与双孔道钾通道介导;钙激活钾通道在胎畜或新生动物中可能具有重要作用,钙激活钾通道与ATP敏感钾通道可调节冠状动脉平滑肌细胞的缺氧性去极化。
电压门控钾通道(Kv)
尽管肺血管表达多种钾通道,电压门控钾通道在膜电位、肺血管张力变化与肺动脉平滑肌细胞增殖中受到广泛关注。电压门控钾通道是最大、多样性最高的钾通道家族,由40个基因编码:36个六跨膜钾通道基因与4个非传导门控调节基因。功能性通道由四个α亚基组成四聚体,每个亚基含六个跨膜结构域,可形成同源或异源四聚体。
急性缺氧时,缺氧刺激可抑制肺动脉平滑肌细胞中电压门控钾通道活性,该效应不仅见于肺动脉平滑肌细胞,也见于缺氧诱导的肺静脉平滑肌细胞收缩。
与之相反,缺氧既不抑制体循环平滑肌细胞的电压门控钾通道活性,也不改变其表达。因此,电压门控钾通道是肺循环的缺氧效应分子,而非体循环平滑肌细胞的效应分子,介导血管收缩。
电压门控钾通道的抑制被认为可由线粒体和/或NADPH氧化酶产生的ROS减少或增多介导。总体而言,半胱氨酸残基的氧化还原修饰对电压门控钾通道活性至关重要。
有研究认为,生理水平ROS触发正反馈机制,降低电压门控钾通道活性;Nox4来源的ROS增多可抑制电压门控钾通道电流;NADPH氧化酶激活与后续H₂O₂生成参与大鼠肺动脉中血栓烷受体激活诱导的电压门控钾通道抑制与收缩反应。
相反,线粒体复合体Ⅰ抑制剂鱼藤酮与黄素蛋白抑制剂二苯基碘鎓(DPI)可抑制缺氧性肺血管收缩与电压门控钾通道电流。但鱼藤酮对线粒体呼吸的作用高度依赖浓度,既可触发缺氧性肺血管收缩,也可抑制肺血管收缩反应。高浓度鱼藤酮诱导的肺血管收缩(程度与缺氧性肺血管收缩相近)被归因于非特异性效应,而非ROS生成改变。线粒体呼吸链近端与远端的其他抑制剂对缺氧性肺血管收缩也表现出相反作用。
急性缺氧通过抑制电压门控钾通道活性发挥作用;慢性缺氧时,钾通道密度与电压门控钾通道蛋白表达(Kv1.5、Kv2.1)降低,但仍可检测到钾电流。慢性缺氧下电压门控钾通道亚基表达既有诱导也有抑制。膜片钳研究显示,环境缺氧2天后缺氧性肺血管收缩已消失,但肺动脉平滑肌细胞中电压门控钾通道电流的缺氧性抑制不变。慢性缺氧3周后,肺动脉平滑肌细胞膜电位去极化,Kv1.5与Kv2.1蛋白降低,急性缺氧对全细胞钾电流的抑制作用消失。另有研究表明,氧化应激(病理水平ROS)下硫氧还蛋白系统慢性受损可导致电压门控钾通道上调。此外,电压门控钾通道还可与吡啶核苷酸相互作用,并发生Cys445的S-亚硝基化修饰。
钙激活钾通道(KCa)
钙激活钾通道按电导分为大电导(BK)、中电导(IK)与小电导(SK),本文重点关注大电导钙激活钾通道。与大电导钙激活钾通道不同,小电导与中电导钙激活钾通道在血管平滑肌细胞中的作用尚不明确。大电导钙激活钾通道在血管平滑肌细胞中广泛表达,可被膜电位与胞内Ca²⁺浓度变化激活,作为血管收缩时去极化与胞质Ca²⁺升高的负反馈机制。胞内Ca²⁺升高可降低大电导钙激活钾通道电流、增加电压门控钾通道电流,因此胞质Ca²⁺不仅是调控这些通道的关键,也对电压变化敏感。
大电导钙激活钾通道存在于肺动脉平滑肌细胞中,但其在全细胞钾电流中的作用具有种属与肺血管树部位差异:近端节段富含大电导钙激活钾通道的肺动脉平滑肌细胞比例更高,远端节段则以电压门控钾通道为主。有观点认为,随肺动脉平滑肌细胞从胎畜、新生到成年成熟,钾通道介导的缺氧反应会发生改变;钙激活钾通道活性在缺氧诱导的胎肺血管舒张中占主导。
大电导钙激活钾通道在缺氧反应中的贡献仍不明确:急性缺氧时,肺动脉平滑肌细胞中大电导钙激活钾通道活性减弱,而肌浆网钙释放可增强其活性。
由于肌浆网钙释放与大电导钙激活钾通道激活及膜超极化相关,急性缺氧介导的钙释放是否触发肺动脉平滑肌细胞膜去极化仍未解决。成年肺动脉平滑肌细胞或肺血管中钙激活钾通道的氧化还原调控研究较少。至少在小鼠肺中,功能性大电导钾通道α亚基敲除不改变急性、持续期缺氧性肺血管收缩与慢性缺氧诱导的肺动脉高压。钙激活钾通道在胎肺缺氧性肺血管收缩中具有重要作用,但这是通过环核苷酸依赖性激酶刺激实现的,最终导致钙激活钾通道激活、膜超极化与血管舒张。
我们认为,ROS对大电导钙激活钾通道活性的作用尚无定论,主要结论来自非肺部研究。图3总结了钙激活钾通道的氧化还原调控假说:线粒体轻度去极化导致ROS升高,激活瞬时钙激活钾电流;线粒体重度去极化则降低ROS,抑制瞬时钙激活钾电流。缺氧可降低钙激活钾通道活性,但其具体作用仍不明确。氧化剂对大电导钙激活钾通道活性的上调或下调依赖实验条件:半胱氨酸氧化降低大电导钙激活钾通道电流,甲硫氨酸氧化则增加电流。钙激活钾通道的氧化还原调控极可能依赖ROS/活性氮(RNS)浓度、氧化剂种类与细胞类型。

图3钙激活钾通道的氧化还原调控假说。
钙激活钾通道的氧化还原调控研究较少。线粒体轻度去极化使ROS升高,激活瞬时钙激活钾电流;线粒体重度去极化则降低ROS,抑制瞬时钙激活钾电流。缺氧可降低钙激活钾通道活性,但其具体作用仍不明确。氧化剂对大电导钙激活钾通道活性的上调或下调依赖实验条件:半胱氨酸氧化降低大电导钙激活钾通道电流,甲硫氨酸氧化则增加电流。钙激活钾通道的氧化还原调控依赖ROS/活性氮浓度、氧化剂种类与细胞类型。?=缺氧对钙激活钾通道的作用尚不明确。EC:细胞外;IC:细胞内;KCa:钙激活钾通道;RNS:活性氮。
ATP敏感钾通道(KATP)
ATP敏感钾通道属于内向整流钾通道亚类,几乎无电压依赖性,基础状态下开放概率低。通道由四个成孔内向整流钾通道亚基(Kir6.1/6.2)与四个磺酰脲受体(SUR)组成八聚体复合物。这些亚基共表达可形成两种不同通道:二磷酸核苷酸敏感钾通道(KNDP)与ATP敏感钾通道。
人肺动脉平滑肌细胞中检测到Kir6.1与SUR2B共表达,提示其参与肺动脉平滑肌细胞静息膜电位调控。相反,ATP敏感钾通道抑制剂不增加成年哺乳动物常氧肺血管阻力,提示此类通道不调控基础肺动脉张力。目前尚无假说认为ATP敏感钾通道参与缺氧性肺血管收缩,其在肺动脉中处于关闭状态,且不被引发收缩的缺氧水平激活。
与钙激活钾通道类似,肺动脉平滑肌细胞中ATP敏感钾通道的氧化还原调控研究较少。总体而言,ATP敏感钾通道由细胞内核苷酸门控,将能量代谢与膜兴奋性关联。肺外组织中越来越多证据表明ATP敏感钾通道活性受氧化还原状态调控,但几乎所有氧化还原依赖性调控机制均来自肺外组织,因此本文不再详述。
双孔道钾通道(K2P)
双孔道钾通道含四个跨膜结构域与两个孔道结构域,两个亚基形成二聚体构成单孔道(共两个孔道),N端与C端位于胞质。双孔道钾通道对K⁺具有选择性,是调控静息膜电位的背景钾通道,调节细胞兴奋性与K⁺通透性。其调控复杂,可响应pH、牵张、温度、脂肪酸、氧张力、小泛素化修饰、磷酸化、去磷酸化与渗透压等多种刺激。由于K⁺选择性与电压非依赖性门控,双孔道钾通道适合介导背景钾电流。
双孔道钾通道包括六个亚家族:TWIK、TREK、TASK、TASK-2、THIK、TRESK。肺血管中已证实表达TASK-1、TASK-2、TREK-2、THIK-1、TWIK-2。但只有TASK-1与TASK-2(主要是TASK-1)参与非失活背景钾电导,缺氧诱导的TASK-1抑制参与肺动脉平滑肌细胞去极化与缺氧性肺血管收缩。
由于TASK-1本身无法感知氧,NADPH氧化酶NOX4被认为是调节其氧敏感性的氧感受分子。在HEK293细胞中,缺氧诱导NOX4激活抑制TASK-1活性,该过程中血红素结构域与FAD结合域参与NOX4对TASK-1的调控。此外,HeLa细胞中TASK-1、TASK-3与TASK-1/3异聚体可被H₂O₂激活,该效应不依赖巯基氧化,提示H₂O₂直接作用于通道蛋白。
相反,TREK-1、TREK-2、TALK-1、TASK-2、TRESK对H₂O₂无反应。黄嘌呤/黄嘌呤氧化酶产生的超氧阴离子仅影响HeLa细胞中TASK-2活性。值得注意的是,H₂O₂浓度至16.3 mM仍无显著效应,该浓度远超生理范围,可能引发非特异性作用。早期研究得出矛盾结果:CHO细胞中表达的TASK-1、TASK-3、TRAAK对H₂O₂无反应,而TREK-2被H₂O₂激活,推测是H₂O₂诱导肌球蛋白轻链激酶(MLCK)激活的结果。该差异可由H₂O₂浓度(5 mM)更低与细胞类型不同解释,并得到其他研究部分支持:缺氧(及ROS)抑制颈动脉体Ⅰ型细胞中TASK-1与TASK-2单通道活性。
钙通道
胞内Ca²⁺浓度是血管张力调控的核心。稳态下胞内Ca²⁺约100 nM,胞外1.6 mM,巨大浓度差凸显细胞Ca²⁺稳态严格调控的重要性。钙通透性通道允许Ca²⁺顺电化学梯度跨膜内流,钙泵逆浓度梯度转运Ca²⁺,钙交换体则可根据作用模式将Ca²⁺转运至胞内或胞外。
调控肺动脉平滑肌细胞胞质Ca²⁺浓度的通道包括:
1. 电压门控钙通道(VOCCs)介导胞外Ca²⁺内流;
2. 受体操纵性阳离子通道(ROCs);
3. 肌浆网耗竭激活的钙池操纵性通道(SOCs)。
胞内Ca²⁺浓度改变在肌肉收缩、细胞运动、神经递质释放、神经元兴奋性、学习记忆、受精发育、细胞增殖、分化、凋亡与基因转录中具有重要作用。Ca²⁺内流对肺前毛细血管缺氧性收缩至关重要。
肺动脉平滑肌细胞中最明确的Ca²⁺内流通路为电压门控钙通道(受静息膜电位调控)与瞬时受体电位通道(TRPC,电压非依赖性非选择性阳离子通道,包括钙池操纵性与受体操纵性通道)。
肺动脉平滑肌细胞胞内Ca²⁺升高是缺氧性肺血管收缩的关键事件已被广泛认可。肺血管中,肺动脉平滑肌细胞急性缺氧钙释放较小程度依赖电压门控钙通道(抑制使缺氧钙释放降低30%),更大程度依赖其他跨膜通道如瞬时受体电位通道(抑制使缺氧钙释放降低60%)。这些通道主要介导缺氧性肺血管收缩相关的持续性肺动脉平滑肌细胞收缩与肺缺氧反应调节。
电压门控钙通道(VOCCs)
肺动脉平滑肌细胞膜电位主要由钾通道调控。缺氧等导致K⁺外流抑制引发细胞去极化,去极化达到阈值后电压门控钙通道激活,即兴奋–收缩耦联。此外,L型通道抑制剂可抑制缺氧性肺血管收缩,这一发现形成了长期假说:缺氧性肺血管收缩主要由氧化还原介导的延迟整流钾通道抑制、去极化与电压依赖性钙内流引起。
电压门控钙通道在血管平滑肌细胞中广泛表达,亚基包括α₁、β₁–β₄、γ₁–γ₈、α₂δ₁–α₂δ₃,每个亚基含六个跨膜结构域(S1–S6),N端与C端位于胞内。多种亚基可共组装,形成电压门控钙通道的异质性。α₁亚基是主要亚基,含Ca²⁺选择性孔道(S5–S6之间)与电压感受器(S4),是通道功能必需的,还包含细胞内第二信使、毒素与药物的调控位点。α₁亚基与不同辅助亚基组合形成六个功能不同的亚家族:L型、N型、P/Q型、R型、T型。这些主要亚型在多种细胞中表达,负责肌肉收缩、动作电位调控、分泌与基因表达等功能。
血管平滑肌细胞中研究最广泛的是二氢吡啶敏感、高电压激活、缓慢失活的L型与低电压激活、快速失活的T型通道。
L型通道在肺动脉平滑肌细胞中研究广泛,在缺氧时细胞Ca²⁺浓度升高中起重要作用。L型通道为高电压激活型,调控兴奋–收缩耦联。慢性缺氧诱导的肺动脉高压中L型通道上调,与钙依赖性阻力相关。与传导动脉相比,阻力动脉肺动脉平滑肌细胞中L型钙通道密度高两倍。
肺血管中另一常见钙通道为低电压激活T型通道。此类通道对常见L型通道阻滞剂不敏感,生理意义尚不明确。肺血管中T型通道的表达与功能研究较少,电生理特性需全面阐明。近期研究提示T型通道在人肺动脉平滑肌细胞增殖中具有重要作用。R型电流可被内皮素-1激活,在蛛网膜下腔出血时增强脑血管收缩。
缺氧增强阻力动脉肺动脉平滑肌细胞Ca²⁺内流,却抑制传导动脉平滑肌细胞Ca²⁺内流(与体循环动脉类似)。如前所述,肺阻力动脉平滑肌细胞急性缺氧钙释放较小程度依赖电压门控钙通道,更大程度依赖瞬时受体电位通道等其他跨膜通道。尽管如此,电压门控钙通道是最早被鉴定为氧化还原敏感的钙通道之一。氧化剂影响电压门控钙通道活性、表达、转运、开放时间与开放概率。成孔α₁亚基中的半胱氨酸残基是ROS的分子靶点,氧化既可激活也可抑制通道活性(图4)。
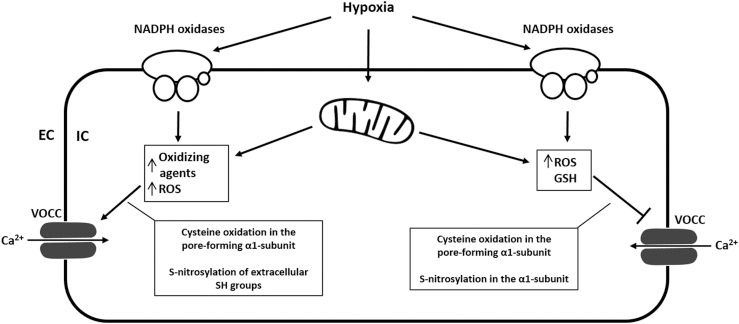
图4电压门控钙通道的氧化还原调控假说。
肺动脉平滑肌细胞去极化达到阈值时电压门控钙通道激活(兴奋–收缩耦联),同时具有氧化还原敏感性。成孔α₁亚基中的半胱氨酸残基是ROS分子靶点,氧化既可激活也可抑制通道活性。氧化剂影响电压门控钙通道活性、表达、转运、开放时间与开放概率。ROS对巯基的氧化降低心肌L型钙电流,而其他氧化剂(DTNB)对巯基的氧化则刺激钙电流。氧化剂对电压门控钙通道的作用依赖种类与作用方式。谷胱甘肽(GSH)抑制电流,已知缺氧时细胞GSH水平降低。L型钙通道胞外巯基S-亚硝基化增加电流,而α₁亚基内S-亚硝基化降低电流。相反结果可由ROS/活性氮的浓度与种类依赖性效应解释。DTNB:5,5′-二硫代双(2-硝基苯甲酸);GSH:谷胱甘肽;SH:巯基。
尽管几乎所有研究均在心肌细胞或心肌L型通道上开展,但有观点认为其他组织细胞的氧化还原调控机制可类推至肺动脉平滑肌细胞电压门控钙通道。但该假设受到以下观察结果挑战:缺氧对阻力动脉肺动脉平滑肌细胞Ca²⁺内流(缺氧增强)与传导动脉平滑肌细胞Ca²⁺内流(缺氧抑制)的作用不同。此外,急性缺氧抑制成孔α₁亚基介导缺氧性血管舒张,但缺氧也可选择性增加PC12细胞与小脑颗粒神经元中L型钙通道。因此,电压门控钙通道的氧化还原调控能否跨组织类推仍存疑。
有研究报道,HEK293细胞中表达的人心肌L型α₁亚基在100 μM H₂O₂处理后电流呈电压依赖性增加,过氧化氢酶处理可降低电流。相反,NADPH氧化酶抑制剂二苯基碘鎓与苯胂氧化物对基础钙电流与缺氧反应均无影响。作者认为内源性H₂O₂生成调控α₁亚基,但H₂O₂水平降低或NADPH氧化酶抑制不参与钙通道的氧依赖性调控。
重组表达系统(稳定表达人L型α₁亚基的HEK293细胞)证实缺氧诱导功能性L型钙通道表达增加,该效应归因于缺氧改变α₁亚基转运。
总体而言,急、慢性缺氧时ROS生成改变,ROS对巯基的氧化降低心肌L型钙电流,而其他氧化剂(DTNB)对巯基的氧化则刺激心室肌细胞钙电流。因此,氧化剂对L型通道的作用依赖种类与作用方式。
钙池操纵性与受体操纵性钙通道
内质网与肌浆网Ca²⁺耗竭可触发Ca²⁺跨质膜内流,即钙池操纵性钙内流,由钙池操纵性通道介导。
除通道残基(尤其是Kv1.5)氧化导致钾通道抑制外,缺氧诱导肺动脉平滑肌细胞去极化还可通过多种其他机制启动,包括钙池释放导致胞质Ca²⁺升高。越来越多证据表明,缺氧诱导肺动脉平滑肌细胞去极化至少部分由钙池操纵性通道激活引起。
配体结合、二酰甘油(DAG)与蛋白激酶C(PKC)激活膜受体可刺激受体操纵性通道,引发Ca²⁺内流与Na⁺外流。钙池操纵性通道介导钙池耗竭或受体操纵性通道激活受体–G蛋白–第二信使通路导致的非选择性阳离子通道Na⁺内流,是肺动脉平滑肌细胞去极化与后续电压门控钙内流的重要初始原因。
总体而言,瞬时受体电位通道可形成功能性受体操纵性与钙池操纵性通道。
与电压门控钙通道类似,瞬时受体电位通道属于六跨膜阳离子通道超家族,但无电压敏感性(电压非依赖)。瞬时受体电位通道为非选择性阳离子通道,但主要携带Ca²⁺。
肺血管中已根据激活机制与N/C端调控结构域鉴定出多种亚型:经典瞬时受体电位通道(TRPC1–TRPC7)、香草素受体相关瞬时受体电位通道(TRPV1–TRPV4)、黑素素相关瞬时受体电位通道(TRPM1–TRPM8)。
血管平滑肌细胞中已检测到10余种瞬时受体电位亚型。但远端肺动脉中TRPC1、TRPC4、TRPC6蛋白表达高于近端肺动脉,与缺氧性肺血管收缩时胞质Ca²⁺浓度变化相关;TRPC1在慢性缺氧诱导肺动脉高压的肺血管重构中起重要作用。TRPC6在肺组织、肺血管平滑肌细胞与内皮细胞中高表达。TRPC6敲除小鼠完全缺失缺氧性肺血管收缩急性期,而持续期无显著改变(图5)。
TRPC6与TRPC3是最早被二酰甘油激活的离子通道。常氧时二酰甘油位于胞质,缺氧时转位至质膜门控TRPC6。
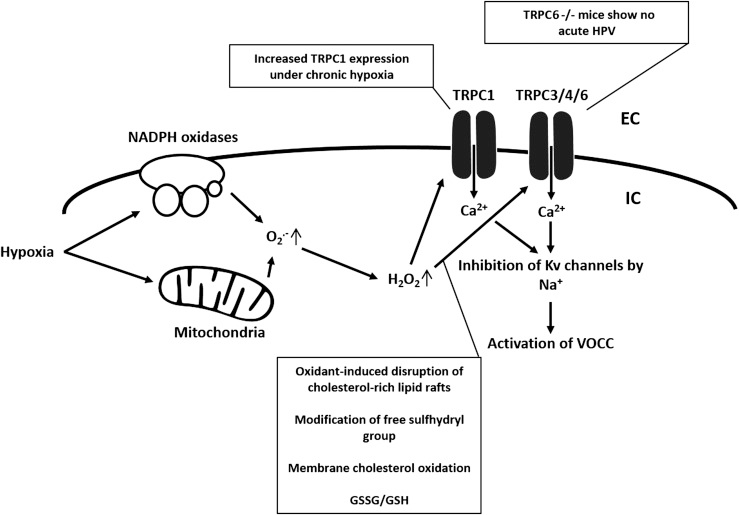
图5含TRPC3/4与TRPC1通道的推测氧化还原调控。
TRPC3/4受ROS调控,含TRPC3/4的通道可在氧化剂存在时激活。ROS诱导富含胆固醇脂筏破坏与膜胆固醇氧化,被认为激活含TRPC3/4的通道。TRPC1在慢性缺氧诱导肺动脉高压的血管重构中起重要作用。TRPC1的氧化还原调控可能与TRPC3/4类似。尽管不能排除磷脂酶C(PLC)与PLC介导膜磷脂酰肌醇磷酸(PIP)水解激活TRPC3/4与TRPC1(类似TRPC6),但氧化应激介导TRPC3激活的机制不涉及PIP水解。PLC在TRPC1激活中的作用尚未阐明。GSSG:氧化型谷胱甘肽。
二酰甘油转位可能由ROS增多触发:肺缺血再灌注(I/R)诱导水肿动物模型研究显示,缺血再灌注缺氧期内皮细胞Nox2来源超氧阴离子生成可激活内皮TRPC6,进而激活磷脂酶C-γ、抑制二酰甘油激酶,最终二酰甘油蓄积并介导TRPC6激活。据此模型,超氧阴离子转化为H₂O₂,通过胞外环触发TRPC6反应(图6)。如前所述,瞬时受体电位通道为非选择性阳离子通道,因此我们进一步推测,TRPC6介导Na⁺内流与肌膜下Na⁺浓度升高可抑制电压门控钾通道、激活L型钙通道(图2)。
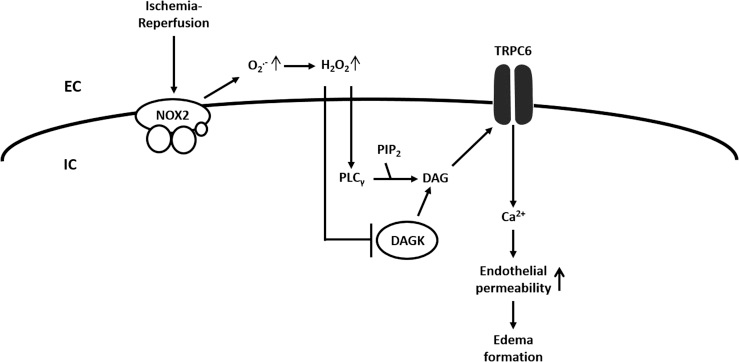
图6瞬时受体电位通道6在肺缺血再灌注损伤中的假说作用。
肺缺血再灌注损伤动物模型中,肺血管内皮细胞TRPC6开放与后续Ca²⁺内流由内皮Nox2来源超氧阴离子生成、磷脂酶C-γ激活、二酰甘油激酶(DAGK)抑制、二酰甘油蓄积与二酰甘油介导TRPC6激活触发。该模型中H₂O₂重新进入细胞(胞外环),激活磷脂酶C-γ并灭活二酰甘油激酶。H₂O₂:过氧化氢;LIRE:肺缺血再灌注诱导水肿;Nox2:NADPH氧化酶2;PIP2:磷脂酰肌醇4,5-二磷酸;PLCγ:磷脂酶C-γ。
已知ROS生成增多可导致胞内钙池钙释放与质膜钙内流。尤其在非兴奋性细胞(如内皮细胞)中,尽管肺微血管内皮细胞表达功能性电压门控T型钙通道,主要钙内流通路仍为钙池操纵性与受体操纵性通道。内皮细胞中TRPC7(TRPM2)与TRPC3/TRPC4受ROS调控,TRPC7已不再归为瞬时受体电位通道家族。含TRPC3/4的通道可被氧化剂激活,参与内皮细胞去极化,并通过氧化剂诱导富含胆固醇脂筏破坏受氧化应激调控。
与TRPC6类似,TRPC3激活受C型磷脂酶与磷脂酶C介导膜磷脂酰肌醇磷酸水解调控。但氧化应激介导TRPC3激活的机制不涉及磷脂酰肌醇磷酸水解。有推测认为,ROS对膜胆固醇的氧化可能是激活TRPC3的信号事件。有研究认为TRPC3与TRPC4构成氧化还原敏感通道的亚基,另外两个亚基尚不明确。磷脂酶C在TRPC1激活中的作用尚未阐明。
钙池操纵性通道也可通过非选择性阳离子通道受细胞氧化还原状态间接调控,该通道可被氧化型谷胱甘肽(GSSG,抗氧化分子)共价修饰。也有观点认为钙池操纵性通道本身可能是氧化型谷胱甘肽(即细胞氧化还原状态)或其他ROS/活性氮的直接靶点。
总体而言,肺中钙池操纵性钙内流通道的氧化还原调控研究较少。急性缺氧(ROS增多)可增强远端肺动脉平滑肌细胞中钙池操纵性通道介导的钙池调控钙内流,引发去极化与电压门控钙通道激活,提示钙池操纵性通道参与缺氧性肺血管收缩。此外,钙池操纵性与电压门控钙通道拮抗剂可抑制缺氧时肺动脉平滑肌细胞收缩与钙池操纵性通道依赖性电压门控钙通道激活。图5给出ROS依赖性TRPC1与TRPC3/4调控假说。
值得注意的是,TRPM2、TRPM7、TRPC5、TRPV1可被ROS激活。TRPC5、TRPV1、TRPA1的激活由游离半胱氨酸巯基氧化触发。这些发现是否适用于肺中其他瞬时受体电位通道仍待验证。
结论
缺氧、缺氧相关疾病及其他影响细胞氧化还原状态的条件下,离子通道表达与门控的氧化还原调控是公认机制。
ROS对离子通道的直接可逆效应包括但不限于:巯基氧化、精氨酸与赖氨酸残基氧化、甲硫氨酸氧化。
间接可逆效应包括:谷胱甘肽水平改变、磷脂酶C(二酰甘油)激活、蛋白激酶C激活、胞质Ca²⁺水平改变。
尽管部分离子通道(电压门控钾通道、瞬时受体电位通道6)的氧化还原调控研究较为深入,但整体仍较为匮乏。
除缺氧时ROS生成增多与减少的争议外,我们还需阐明为何不同氧化剂在不同条件下既可激活也可抑制通道活性。现有文献表明,同一来源的ROS可作用于离子通道的不同氨基酸残基,介导通道开放或关闭。
目前,多数离子通道氧化还原调控机制基于少数已知机制的推测与外推。尽管影响通道门控的上游通路已有较多阐释,但ROS对通路中单个蛋白的作用仍大多未知。
关键问题是更深入理解氧化还原蛋白修饰,以阐明离子通道氧化还原调控的复杂性。阐明ROS的时空协调方式及ROS空间分布的作用至关重要。
显然,生理氧化还原信号在亚细胞区室与微域中具有时空限制性。细胞氧化还原状态并非氧化剂与还原分子的整体失衡,而是不同细胞区室氧化还原状态的净效应。
线粒体作为细胞内氧化还原活性最高的区室,是ROS生成的重要位点。尽管线粒体抗氧化能力极强,过量ROS释放可引发多种疾病。胞质也可作为亚细胞区室,质膜刺激可触发胞质特定蛋白氧化而不影响其他细胞器。
其他氧化还原活性区室包括细胞核、内质网腔、过氧化物酶体、内体与溶酶体。这些区室内ROS微域生成具有高度多样性。
在此背景下,NADPH氧化酶作为另一重要细胞ROS来源,已被证实表达于此类ROS微域(如小窝、内体)。甚至有观点认为,单一NADPH氧化酶亚型(Nox1)可通过占据细胞内不同微域产生多种信号效应。
https://blog.sciencenet.cn/blog-41174-1524613.html
上一篇:氢气吸入对脑血流的影响大【新进展】
下一篇:氢气作为高血压辅助治疗有效性与安全性【真实世界研究】
