博文
药物研发:抗菌药物药效(PD)药代(PK)的给药逻辑_MCE(MedChemExpress)
|
Section.01
药物代谢动力学 (PK)
药物代谢动力学 (Pharmacokinetics, PK) :简称药代动力学,是定量研究药物在生物体内吸收 (Absorption) 、分布 (Distribution) 、代谢 (Metabolism) 和排泄 (Excretion) ,随时间变化规律的学科。
——简单理解,PK 探究:在药物摄入后,机体是如何处理药物的?核心是测定药物浓度随时间的变化。
知识链接
1. 药物的体内动力学过程
• 动力学本身分为零级动力学 (定值) 和一级动力学 (线性动力学) 。
• 目前临床上应用的大多数小分子药物,其体内的吸收、分布、代谢、排泄 (ADME) 过程都遵循一级动力学特征 (线性动力学特征) 。
2. 给药方式
• 血管内给药:如静脉注射、静脉滴注
• 血管外给药:如口服、肌内注射、吸入给药、透皮给药
如何探究?药代动力学通过非房室模型或房室模型进行建模分析。房室模型 (Compartment Model) 使用动力学模型估算血药浓度-时间图。被更广泛地使用的是——非房室模型 (Noncompartmental Analysis, NCA) ,通过估算血药浓度-时间图中的曲线下的面积,来估计药物暴露量。
血药浓度-时间曲线下面积 (AUC)
首先,了解下血药浓度,是指药物吸收后在血浆内的总浓度,这包括了与血浆蛋白结合的药物或在血浆中游离的药物,有时也可泛指药物在全血中的浓度。
血药浓度-时间曲线下面积 (Area under curve,AUC) 指药物在血液中的浓度随时间变化曲线 (药-时曲线) 和横坐标所包围的面积,其数值反映在某段时间内进入人体循环的药量,即药物的吸收程度。
——评价药物吸收程度的重要指标。
药物暴露量:直接反映药物在体内被吸收和暴露的总量。AUC 越大,代表药物在体内停留的时间越长、浓度越高。
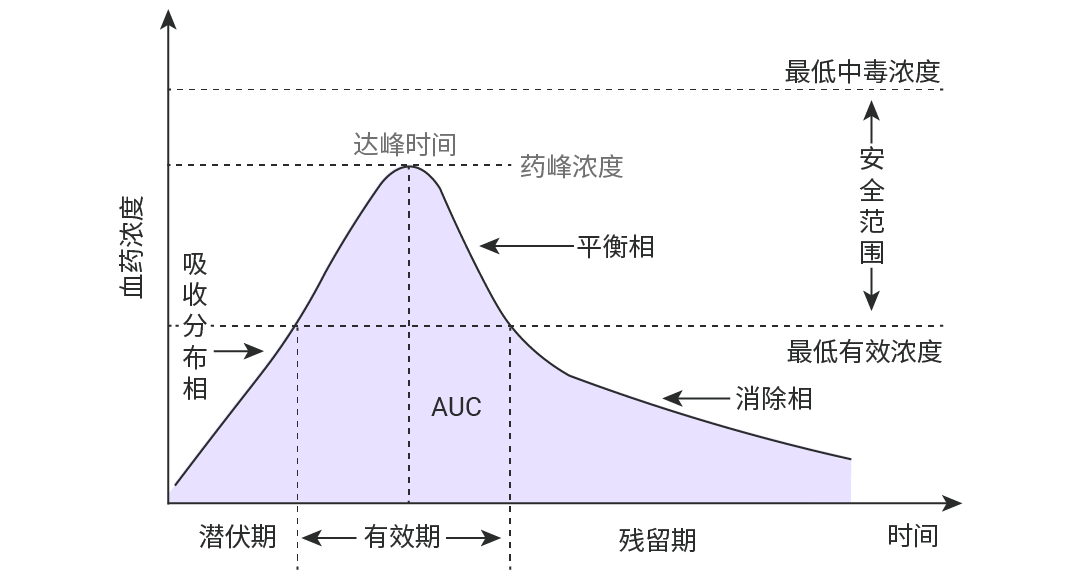 图 1. 单次口服给药后的药-时曲线示意图。
图 1. 单次口服给药后的药-时曲线示意图。
该曲线以时间为横坐标,血药浓度为纵坐标。
AUC0→t表示从给药开始到t时刻的药-时曲线下面积;
AUC0→∞则指从给药开始到所有原形药物全部消除为止时的曲线下总面积。
可通过梯形法则 (数值积分) 计算各单位间隔时间内面积之和 (最常用的手段) 。取样时间点 (采样点) 越密集,预测的梯形面积越能反映浓度-时间曲线的实际情况。
采样点
采样点的确定对药代动力学研究结果有重大影响,若采样点过少或选择不当,得到的血药浓度-时间曲线可能与药物在体内的真实情况产生较大差异。
① 给药前需要采血作为空白样品。
② 为获得给药后的一个完整的血药浓度-时间曲线,采样时间点的设计应兼顾药物的吸收相、平衡相 (峰浓度附近) 和消除相。
③ 一般在吸收相至少需要 2~3 个采样点,对于吸收快的血管外给药的药物,应尽量避免第一个点是峰浓度 (Cmax);在 Cmax 附近至少需要 3 个采样点;消除相需要 4-6 个采样点。
④ 整个采样时间至少应持续到 3-5 个半衰期,或持续到血药浓度为 Cmax 的 1/10-1/20。
⑤ 为保证最佳采样点,建议在正式试验前选择 2~3 只动物进行预试验,然后根据预试验的结果,审核并修正原设计的采样点。
根据试验中测得的各受试动物的血药浓度-时间数据,可求得受试药物的主要药代动力学参数。
例如:静脉注射给药,应提供 t1/2、Vd、 AUC、Cl 等参数值;血管外给药,除提供上述参数外,还应提供 Cmax 和 Tmax 等参数,以反映药物吸收的规律。另外,提供统计矩参数,如: MRT、AUC0→t 和 AUC0→∞ 等,对于描述药物药代动力学特征也是有意义的[1]。
Cmax (药峰浓度) 和 Tmax (药物达峰时间)
药峰浓度,代表符号是 Cmax,是指血管外给药吸收后所能达到的最高血浆药物浓度,即血药浓度—时间曲线 (药-时曲线) 上的最大血药浓度值。单位以 μg/mL 或 mg/L 表示。当 Cmax 达到一定浓度药物才能显效,浓度越高效果越强,但若超出了安全范围则可显示出毒性反应。
药物达峰时间,代表符号是 Tmax,是指机体在给药后,血液中的药物浓度从零开始上升,直至达到最高点 Cmax 时所需的时间。Tmax 是反映药物被身体吸收快慢的重要指标,被作为药物起效的标志。Tmax 数值越小,起效越快,可被用于指导用药间隔,是判断药物起效时间和设计给药频次的重要依据。
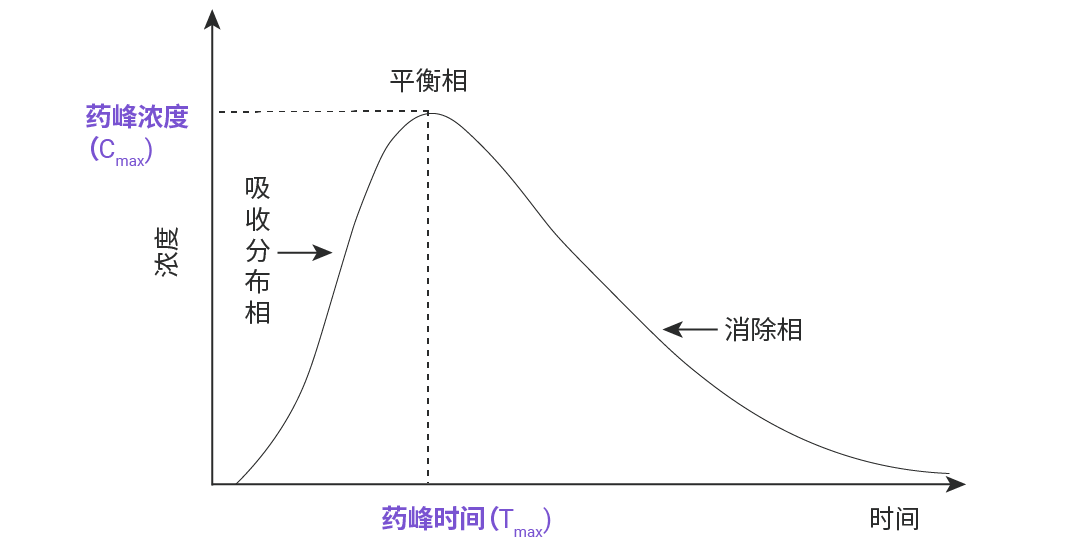 图 2. 血管外给药的药-时曲线。
图 2. 血管外给药的药-时曲线。
——反映药物在体内吸收速率的两个重要指标,药物的吸收速度快,则其峰浓度高,达峰时间短。如图 3 所示,图中 A、B、C 三个制剂的吸收程度相似,但吸收速度不同,其中吸收速度 A>B>C。
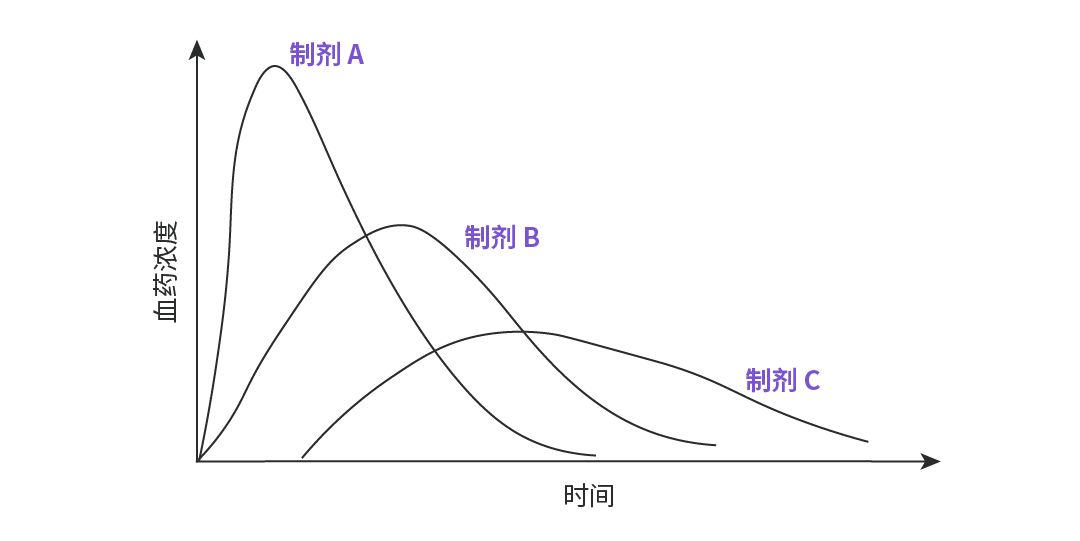 图 3. 制剂 A、B、C 给药后的药-时曲线。
图 3. 制剂 A、B、C 给药后的药-时曲线。
t1/2 (半衰期)
清除半衰期,代表符号是 t1/2,是指血药药物浓度下降一半所需的时间。与消除速率常数 (elimination rate constant, k) 存在倒数关系,公式为:t1/2=0.693/k 。
由于 t1/2更直观,所以临床上多用 t1/2 来反映药物消除的快慢,它是临床确定给药间隔长短的重要依据之一。一般情况下,经过 5-6 个 t1/2 达到稳态血药浓度,药物可基本消除干净。
——t1/2 反映药物消除快慢的程度及机体消除药物的能力。
特征参数:不因药物剂型、给药途径或剂量而改变。
Vd (表观分布容积)
表观分布容积,代表符号是 Vd,是指药物在体内按血中同样浓度分布所需体液的总容积,单位是 L 或 L/kg。
Vd 值的大小反映药物在体内分布的广泛浓度以及组织结合程度,Vd 值并不代表真正的生理体积,其意义在于反映药物的体内分布范围。
Vd 值在不同的药代动力学模型中不同:
非房室模型 (NCA) 中,Vd 通常会细分为两种最常用的计算指标:消除相分布容积 (Vz) 和稳态分布容积 (Vss) 。
对于单室模型的药物而言,分布容积与体内药量 X 和血药浓度 C 之间存在下列关系: Vd=X/C。
药物的分布容积的大小取决于其脂溶性、膜通透性、组织分配系数及药物与血浆蛋白等生物物质的结合率等因素。如药物的血浆蛋白结合率高,则其组织分布较少,血药浓度高。
我们可以根据体液的分布情况 (见表 1),由药物的分布容积可以粗略地推测其在体内的大致分布情况。
表 1. 体液的分布情况。

【以单室模型为例】
① 若一个药物的 Vd 为 3-5 L 左右,那么这个药物可能主要分布于血液并与血浆蛋白大量结合,如双香豆素、苯妥英钠和保泰松等;
② 若一个药物的 Vd 为 10-20 L 左右,则说明这个药物主要分布于血浆和细胞外液,这类药物往往不易通过细胞膜,因此无法进入细胞内液,如溴化物和碘化物等;
③ 若一个药物的分布容积为 40 L,则这个药物可以分布于血浆和细胞内、外液,表明其在体内的分布较广,如安替比林;
④ 有些药物的 Vd 非常大,可以达到 100 L 以上,这一体积已远远地超过了体液的总容积,这类药物在体内往往有特异性的组织分布,如硫喷妥钠具有较高的脂溶性,可以大量地分布于脂肪组织,而 I131 可以大量地浓集于甲状腺,因而其分布容积也很大。
——由此可见我们可以通过分布容积来了解药物在体内的分布情况。
Cl (清除率)
清除率,代表符号是 Cl,是指单位时间内,机体能将多少容积含药血浆从体内清除,单位是 L/h 或 L/h/kg,表示从血中清除药物的速率或效率。
——反映药物从体内消除的一个重要参数。
清除率与清除速率常数k和分布容积之间的关系可用公式:Cl=k · vd 表示。
产品推荐
MTT 是一种黄色的水溶性四唑盐,活细胞线粒体中的脱氢酶可以将 MTT 还原成不溶性的紫色结晶甲瓒,通过测量甲瓒的颜色变化来反映细胞活力。
WST-8 在电子载体 1-Methoxy PMS 的作用下,可以被细胞内脱氢酶还原成水溶性的橙黄色甲臜 (formazan)。
WST-1 诱导细胞内线粒体脱氢酶进行 NADH 依赖的酶切反应,释放出水溶性的甲臜产物,通过测定 450 nm 处的吸光值来反映细胞活力。
参考文献
[1]化学药物非临床药代动力学研究技术指导原则
[2]Zaman A, et al. Quantitative Framework for Bench-to-Bedside Cancer Research. Cancers (Basel). 2022 Oct 26;14(21):5254.
[3]Gui, H., et al. Effects of Boletus Poisoning on Estrogen Receptors and Neurotransmitters in Rats Based on ERk1/2 Pathway. Neural Process Lett 55, 193–203 (2023).
[4]Abal P, et al. Acute Oral Toxicity of Tetrodotoxin in Mice: Determination of Lethal Dose 50 (LD50) and No Observed Adverse Effect Level (NOAEL). Toxins (Basel). 2017 Feb 24;9(3):75.
[5]Ikenoya M, et al. Y. Inhibition of rho-kinase-induced myristoylated alanine-rich C kinase substrate (MARCKS) phosphorylation in human neuronal cells by H-1152, a novel and specific Rho-kinase inhibitor. J Neurochem. 2002 Apr;81(1):9-16.
[6]Wang Z.H, et al. High-afnity SOAT1 ligands remodeled cholesterol metabolism program to inhibit tumor growth. BMC Medicine. 2022 Aug 9;20(1):292.
[7]抗菌药物药代动力学/药效学研究技术指导原则
https://blog.sciencenet.cn/blog-3536222-1538151.html
上一篇:Aβ蛋白与阿尔茨海默症_MCE(MedChemExpress)
