博文
极具潜力的癌症分子标志物:DNA甲基化
||
极具潜力的癌症分子标志物:DNA甲基化
临床上,原发性肝细胞癌(hepatocellularcarcinoma)目前仅有两个常用的分子标志物AFP(甲胎蛋白)和SF(铁蛋白),均是上世纪60、70年代发现的。在找肝癌分子标志物的相关研究中,很大一部分都是找基于遗传物质的分子标志物,但是很难将这些分子标志物与临床的预后及治疗结合起来。
DNA甲基化异常是肿瘤发生发展过程中的标志性事件之一。人类基因启动子区的CpG岛通常是非甲基化的状态,在癌症中CpG岛会发生明显的高甲基化现象(CpG island methylator phenotype,CIMP),可能会导致一些重要的抑癌基因、DNA修复基因的转录沉默,同时全基因通常呈现出去甲基化的状态(genome-wide hypo-methylation),与基因组的稳定性有很大关联。这两种异常变化均与肿瘤的发生发展密切相关。2015年Villanueva A(Hepatology 2015)等人对肝细胞癌患者异常甲基化的研究显示,68%的探针存在低甲基化,32%的探针存在高甲基化的现象,且这些高甲基化的探针大部分都位于Promoter区的CpG岛上。
为了进一步研究肝癌DNA异常甲基化的情况,我们首先用基于分位数的方法分析了TCGA的50个肝细胞癌患者的癌组织和癌旁的全基因组DNA甲基化数据,可以明显的观察到上述两种肝癌DNA异常甲基化的现象(图1,左图为全基因组甲基化水平中值,可观察到显著的全基因组去甲基化现象;右图为CpG岛甲基化高四分位值,可观察到显著的CIMP现象)。

图1 TCGA样本全基因组及CpG岛甲基化状态
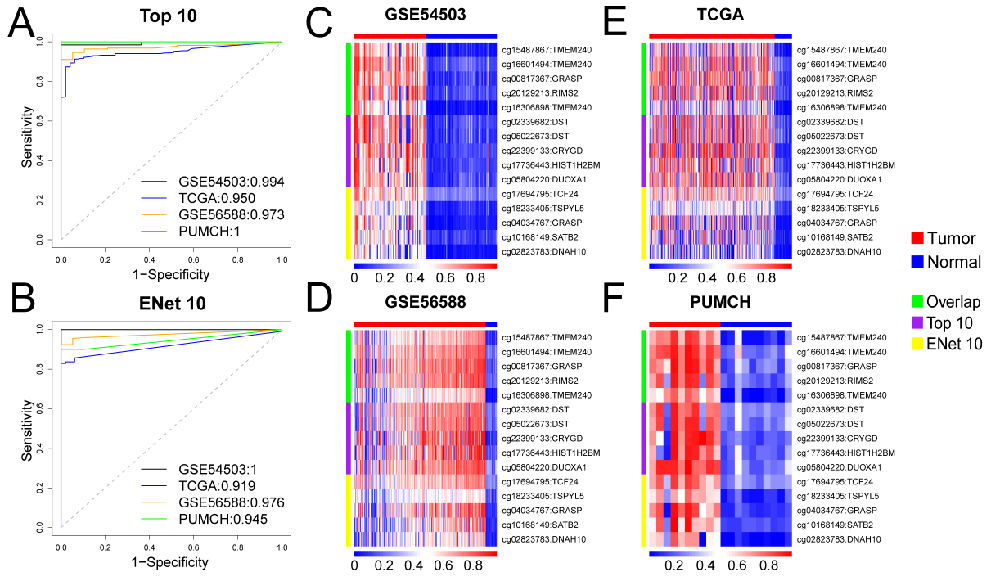
随后,我们广泛收集了公开发表的三组DNA甲基化数据集,并与北京协和医院合作新检测了一些临床样本,共计646个tumor样本和134个non-tumor样本的甲基化数据,令人兴奋的是,仅提取以上两个特征对tumor/non-tumor构建分类器,就实现了很好的分类效果(如下图)!

我们接下来对所收集样本的甲基化数据和基因表达数据进行了系统的整合分析,结果表明:肝细胞癌患者基因启动子区的少数高甲基化位点即可区分癌和非癌组织的分子标志物(如下图)。这些结果表明,DNA甲基化是肝癌极好的候选生物标志物,其应用前景还需在血液检测中进一步进行验证。
同时,我们的分析还发现:
>>> 通过对DNA甲基化数据及基因表达数据的相关性分析,找到了一些与免疫反应及代谢过程相关的表观遗传分子标志物;
>>> 从生存期分析的结果来看,SFN, SPP1 和TKT等基因与肝癌病人的预后密切相关。
(作者:黄倩倩 修改:古槿)
数据1:http://bioinfo.au.tsinghua.edu.cn/member/jgu/hcc-dnameth
数据2:HCCdb http://bioinfo.au.tsinghua.edu.cn/database/hccdb
Briefings in Bioinformatics 2016, Advanced Access
Genome-wide DNA methylation analysisidentifies candidate epigenetic markers and drivers of hepatocellular carcinoma
Yongchang Zheng1,*, Qianqian Huang2,*,Zijian Ding2, Tingting Liu3, Chenghai Xue3,4, Xinting Sang1, Jin Gu2,#
The alteration of DNA methylation landscapeis a key epigenetic event in cancer. As the accumulation of large-scalegenome-wide DNA methylation data from clinical samples, we are able tocharacterize the patterns of DNA methylation alterations for identifyingcandidate epigenetic markers and drivers. In this survey, we takehepatocellular carcinoma (HCC) as an example to show the basic steps ofanalyzing the DNA methylation patterns in cancer across multiple datasets. Wecollected three genome-wide DNA methylation datasets with ~800 clinical samplesand the corresponding gene expression datasets. Firstly, by quantitativelyanalyzing two global methylation alterations, it is found that about 90% tumorsacquire either genome-wide DNA hypo-methylation (GDH) or CpG island methylatorphenotype (CIMP). Secondly, probe-level analysis identified 267, 228 and 197hyper-methylated sites in promoter regions for the three datasets,respectively. These local hyper-methylated patterns are highly consistent: 84sites (from 61 promoters) are hyper-methylated in all the three studieddatasets, including many previously reported genes, such as CDKL2, TBX15 andNKX6-2. Then, these hyper-methylated sites were used as candidate markers toclassify tumor and non-tumor samples. The classifiers based on only 10 selectedprobes can achieve high discriminative ability across different datasets.Finally, by integrative analyzing DNA methylation and gene expression data, weidentified 222 candidate epigenetic drivers, which are enriched in inflammatoryresponse and multiple metabolic pathways. A set of high-confidence candidates,including SFN, SPP1 and TKT, are significantly associated with patients’overall survivals. In summary, this study systematically characterized the DNAmethylation alterations and their impacts on gene expressions in HCCs based onmultiple datasets.
https://blog.sciencenet.cn/blog-407531-1012521.html
上一篇:交叉学科生物信息学的三类研究
下一篇:当肿瘤遭遇“信息泄露”