博文
Cell Metabolism :饿肚子不用怕,SAMe保护你
||
代谢学人
Cell Metabolism :饿肚子不用怕,SAMe保护你
撰文 | 夏志蕊 闪光余 朱丽君 刘爽 曹玉香
编辑 | 孟美瑶
校对 | 朱丽君

背景介绍
禁食可以促进机体健康、延缓衰老、防止肥胖,对人体有益;同时禁食可以触发如肝脏、大脑和脂肪组织等主要器官系统的适应性代谢反应,这是由激素、生长因子和细胞因子的复杂网络控制的。在大多数哺乳动物肝脏中,12-24小时的禁食会促使机体将肝脏储存的糖原、脂肪衍生的酮体和游离脂肪酸作为生命活动的主要能量来源。这种肝脏代谢的适应非常复杂,受到严格的调控,包括葡萄糖、脂质和蛋白质代谢的变化。
目前有研究认为,蛋氨酸(Met)途径中的酶可能在禁食期间被调节以维持蛋氨酸的水平(小编注:蛋氨酸是构成人体的必需氨基酸之一,又名甲硫氨酸,能够参与蛋白质的合成。在绝大多数生物体内,蛋白质合成并不是从RNA分子任意碱基起始的,而是从一个起始密码子开始,起始密码子AUG在真核系统中编码甲硫氨酸,在原核系统中编码甲酰甲硫氨酸,缺乏甲硫氨酸将阻滞基因表达和蛋白质的合成。并且蛋氨酸主要在机体内参与转甲基化反应,转硫反应和多胺合成。在转甲基化反应中,SAM的甲基被转移到多种受体底物,包括DNA,磷脂和蛋白质,因此会影响从基因表达到膜流动性的广泛过程。在转硫过程中,SAM的硫原子通过一系列酶促步骤转化为半胱氨酸,半胱氨酸是牛磺酸和谷胱甘肽的前体,谷胱甘肽是一种主要的细胞抗氧化剂。蛋氨酸参与合成的多胺是正常细胞生长所必需的。因此在生物体中维持蛋氨酸的水平对于生物体有着重要的意义。)蛋氨酸在肝脏中通过蛋氨酸腺苷转移酶(MAT)同工酶I和III代谢合成S-腺苷蛋氨酸(SAMe)。蛋氨酸腺苷转移同工酶主要有三种:MATI、MATII、MATIII。MATI和III的催化亚基主要由在肝脏中表达的MAT1A基因编码,而MATII的催化亚基由MAT2A基因编码。在肝脏中,SAMe有助于多胺的生物合成,参与谷胱甘肽(GSH)的合成以及转甲基化反应。特别是在由磷脂酰乙醇胺-N-甲基转移酶(PEMT)(小编注:磷脂酰乙醇胺-N-甲基转移酶是一种重要的酶类,其主要作用是将磷脂乙醇胺(PE)转化为脂乙醇胺甲基化产物(MPE),从而调节细胞膜的组成和功能)催化的反应中,SAMe还可以作为甲基供体将磷脂酰乙醇胺(PE)转化为磷脂酰胆碱(PC)。
目前,已有研究表明Met循环在维持脂质稳态中的重要作用。蛋氨酸限制饮食(MRD)的啮齿动物表现出肝脏Met和SAMe水平降低,寿命延长,体重增加减缓慢,胰岛素敏感性增加,并且不会发生肝脏脂肪变性。MRD还可以预防小鼠高脂饮食诱导的肥胖、2型糖尿病和肝脂肪变性。在人类中,血浆SAMe水平随着体重指数的增加而增加,而在体重增加较多的人群中,也发现SAMe(但不包括Met)与躯干肥胖和脂肪量相关。另一方面,在禁食期间,肝脏SAMe水平下降,而Met水平保持不变,这表明饥饿状态下肝脏SAMe的利用增强。综上所述,这些结果表明SAMe水平变化(而非Met)可以调节进食/禁食状态下的脂质稳态。然而,基本代谢物SAMe在机体对禁食的代谢反应中变化的功能重要性在很大程度上仍未被探索。
近期发表在Cell Metabolism杂志上的一篇文章“Hepatic levels of S-adenosylmethionine regulate the adaptive response to fasting”揭示了禁食期间肝脏SAMe水平的下降可以通过微调肝脏中线粒体相关膜(MAMs)(小编注:线粒体相关ER膜 (MAMs) 是内质网膜的一个功能子域,它与线粒体外膜相连,对细胞稳态至关重要)的相互作用、β氧化和ATP合成而作为营养代谢传感器。值得注意的是,研究人员观察到肝脏SAMe水平的降低诱导成纤维细胞生长因子21 (FGF21)启动子的低甲基化,增强其合成和分泌,促进腹股沟白色和棕色脂肪组织(ingWAT和BAT)的脂质分解代谢,增加能量消耗(EE)。科研人员通过代谢组学分析表明,在禁食期间,Met循环流向SAMe和PC合成。虽然SAMe的整体水平下降,但禁食期间MAMs中MAT1A蛋白的表达增加,这促进了SAMe生成。这些数据表明,在MAMs中,这种局部合成SAMe在维持禁食期间PEMT活性和PC/PE比率中具有重要作用,这限制了线粒体β氧化和ATP产生的速度,最终防止内质网应激(ERS)和肝损伤。总之,该研究团队的工作揭示了肝脏SAMe在对禁食的适应性代谢反应中的关键作用。

敲黑板啦!
1.营养限制可以提高肝脏中SAMe的产量,并增加SAMe的利用;
2.在禁食期间的肝脏中,SAMe通过维持PEMT活性,来抑制内质网-线粒体的接触及线粒体β氧化和ATP的产生;
3.肝脏SAMe水平降低,增加FGF21循环水平并促进脂质分解代谢,增加能量消耗;
4.肝脏SAMe水平降低,增加线粒体与内质网的相互作用;
5.MAT1A定位于MAMs调节局部SAMe合成,限制线粒体β氧化和ATP产生,从而减轻ERS和肝损伤。
研究结果
1.营养应激增加肝脏中SAMe的利用
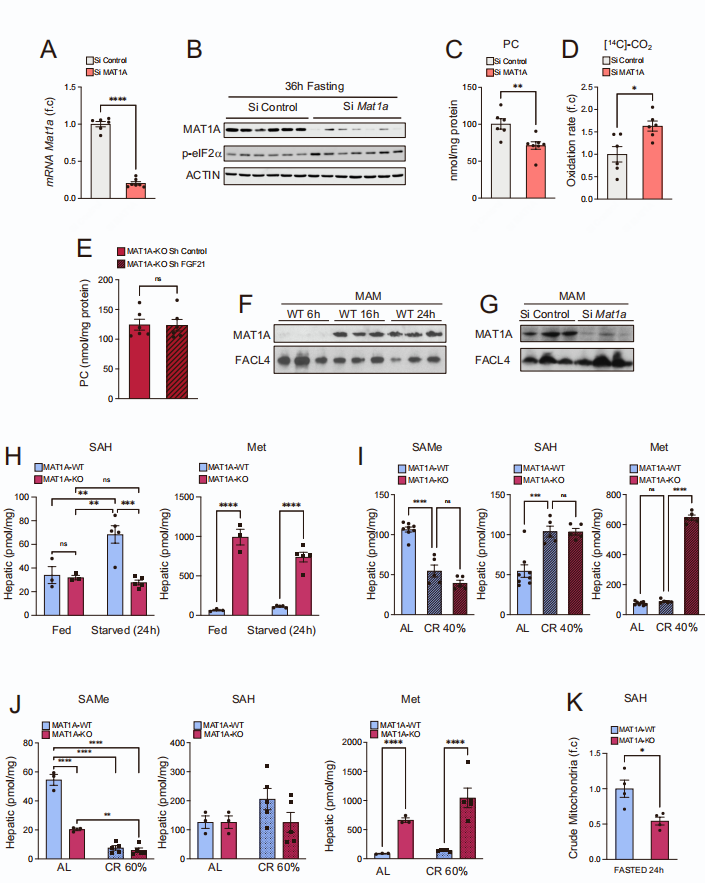
先前有报道称禁食16小时后肝脏中SAMe的水平下降,因此研究人员对禁食后肝脏Met、SAMe和S-腺苷-L-高半胱氨酸(SAH)(小编注:SAH是一种氨基酸衍生物,在大多数生物体中的一些代谢途径中有着重要作用,是合成半胱氨酸和腺苷的代谢中间物,由S-腺苷-L-甲硫氨酸(SAM)脱甲基形成)水平进行了详细的时间过程研究,发现肝脏SAMe水平早在禁食后6小时就显著下降。但SAMe水平的下降是可逆的,因为在禁食24小时后,2小时的再喂养足以部分恢复SAMe水平。另一方面,甲基从SAMe转移后形成的副产物SAH的水平增加,导致SAMe/SAH比率降低(图1A)。值得注意的是,当小鼠在5天内保持60%的热量限制(CR)时发现类似的结果,包括肝脏SAMe水平和SAMe/SAH比率显着降低(图1B)。由于肝脏SAMe主要由MAT1A合成,研究人员在禁食后分析了Mat1a mRNA和MAT1A蛋白水平,并发现Mat1a mRNA和MAT1A蛋白的表达从12小时到24小时逐渐增加,并在36小时持续升高(图1C、D)。同样,Mat1a mRNA和MAT1A蛋白水平在5天的60% CR期间(图1E)、2个月40%CR或隔日禁食期间(ADF)也显著增加(辅图1A、B)。
糖皮质激素,如曲安奈德酮(triamcinolone)和地塞米松(dexamethasone),先前已被证明可诱导Mat1a mRNA的表达。因此,研究人员研究了胰高血糖素是否也可以调节Mat1a mRNA水平(小编注:糖皮质激素和胰高血糖素均可升高血糖,在体内具有协同作用。可升高血糖的激素有(1)胰高血糖素:由胰岛细胞分泌,促进肝糖原分解和减少葡萄糖的利用;(2)肾上腺素:由肾上腺髓质分泌,促进肝糖原的分解和肌糖原的醇解;(3)糖皮质激素:由肾上腺分泌的一组类固醇激素,通过抑制肌肉和脂肪组织摄取葡萄糖,促进肝脏中的异生来提高血糖;(4)生长激素:由垂体后叶分泌,对抗胰岛素;(5)甲状腺激素:促进小肠黏膜对葡萄糖的吸收,同时促进糖原分解及糖异生作用,从而升高血糖。),并发现在胰高血糖素处理12和16小时后,原代肝细胞中Mat1a mRNA和蛋白水平分别上调(图1F)。用胰高血糖素治疗过夜禁食野生型(WT)小鼠仅30分钟就导致体内肝脏Mat1a mRNA水平轻微升高,而在缺乏胰高血糖素受体的小鼠(Gcgr-KO小鼠)中,这种作用被消除(图1G)。这表明空腹期间Mat1a mRNA的上调可由胰高血糖素控制。总之,以上研究结果表明,营养限制诱导了负责SAMe合成的酶上调,同时导致肝脏SAMe水平的降低和其副产物SAH水平的提高。这表明,营养限制可以提高肝脏中SAMe的产量,并随之增加SAMe的利用。
拓展阅读
热量限制(caloric restriction,CR)
热量限制(CR)是指在提供充分的营养成分如必需氨基酸、维生素等,保证不发生营养不良的情况下,限制每日摄取的总热量,将总热量减少25-50%。有研究表明,热量限制不但能延缓肿瘤生长,还能延长实验动物的寿命。30%的CR且不限制禁食时间时,小鼠延长了约10%的寿命;而当给予30%CR的小鼠被限制仅在夜晚进食(小鼠一般在晚上更加活跃)时,能够延长小鼠35%的寿命。表明除了减少每日能量摄入外,CR还重置了整个身体组织中基因表达的复杂昼夜节律程序。虽然减少能量摄入通常被认为是延长寿命的关键因素,但食物摄入的时间可能也是一个重要因素。
限时喂养引起的变化对生理学产生深远的影响。例如,只在白天喂食高脂肪饮食的小鼠比只在夜间喂食相同饮食的小鼠体重明显增加。此外,与自由进食的小鼠相比,在夜间进食限制在8小时的高脂肪饮食的小鼠可防止饮食诱导的肥胖、肝脂肪、高胰岛素血症和炎症。因此,食物摄入时间的选择可能也是延长寿命并改善代谢的重要因素。
目前,对于CR延缓生物衰老和预防老年相关疾病的研究结果较为突出,但是使人们忽略了CR对健康可能带来的不利影响,虽然这方面的研究资料相对较少,但已有的研究表明不适当CR可引起多种健康问题如低血压、体重过低、性欲散失、女性月经不调、生育能力减退、骨质疏松等。
参考文献
[1] Acosta-Rodriguez, et al. Science, 2022. 376(6598):1192-1202
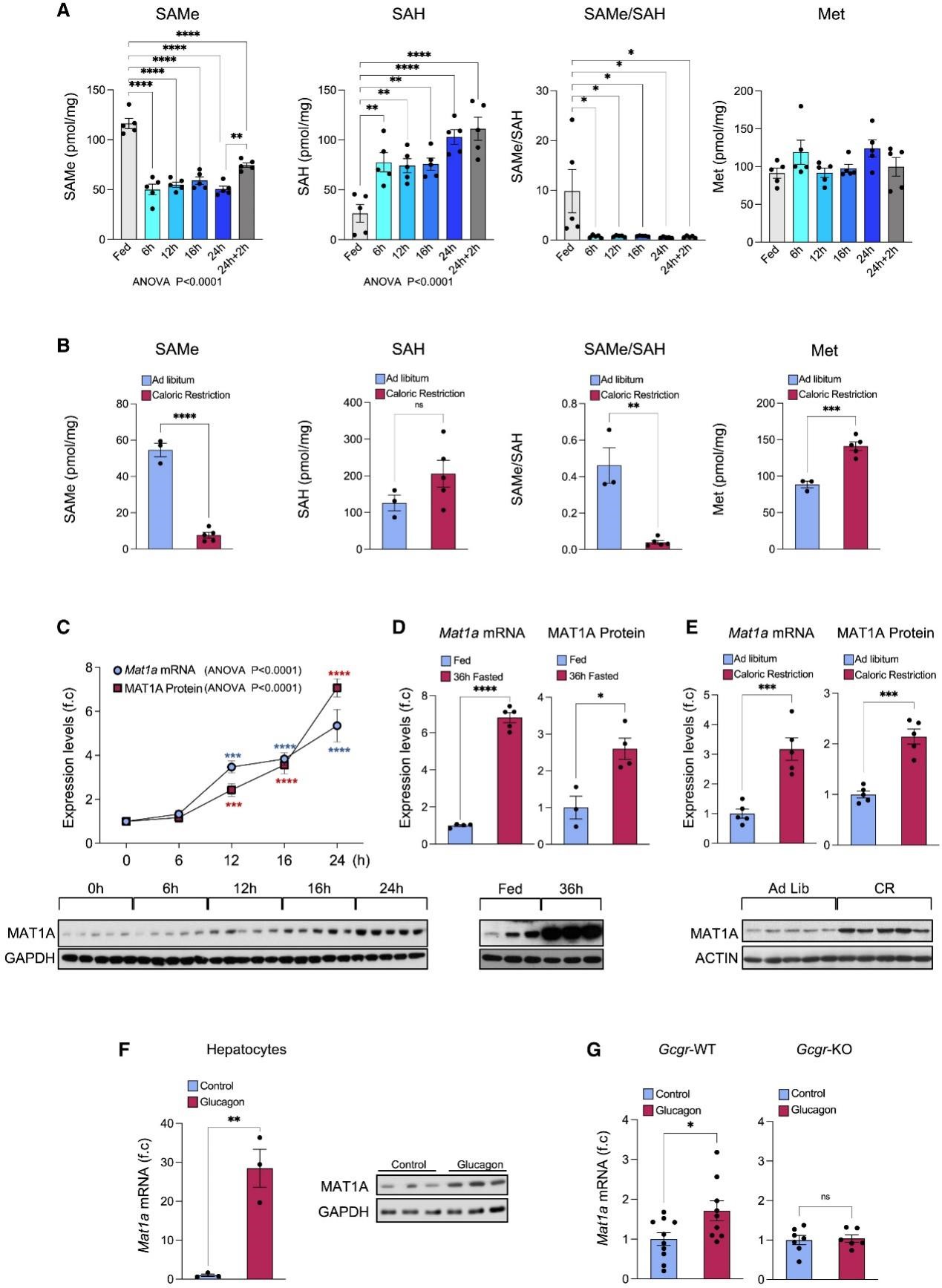
图1 营养应激导致肝脏SAMe利用增加
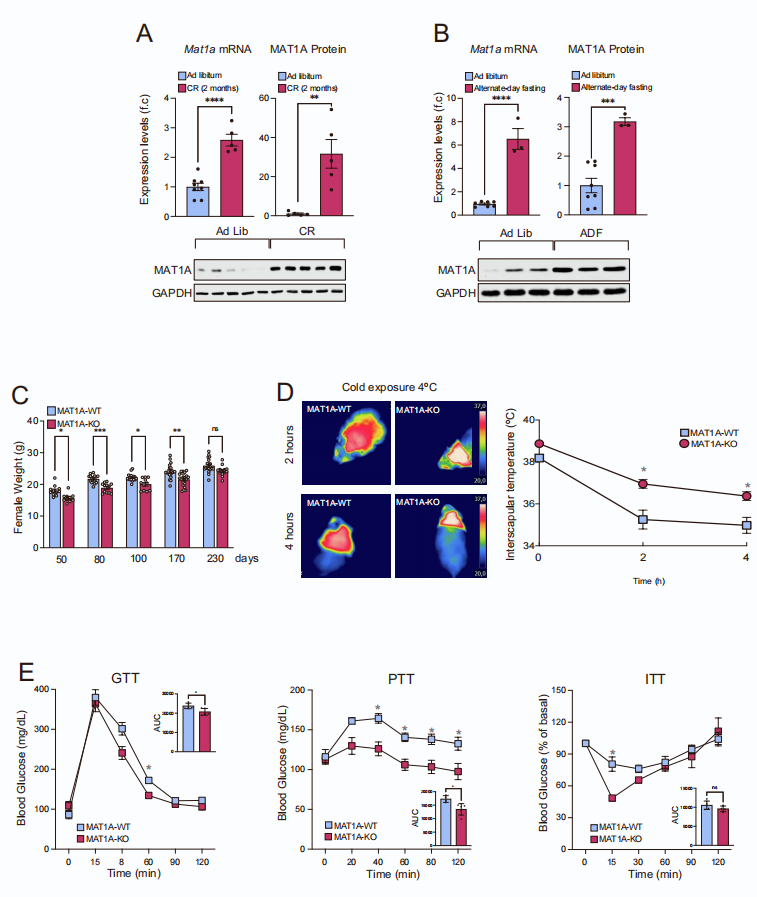
辅图1 禁食诱导上调MAT1A的表达,增加能量消耗并增加葡萄糖和丙酮酸耐量、改善胰岛素耐敏感性
2.MAT1A-KO小鼠出生后死亡率增加且体重增量减缓
接下来,研究人员利用敲除MAT1A的小鼠模型(MAT1A-KO小鼠)探索肝脏MAT1A在脂质代谢中的作用。这些小鼠的特点是显著的高蛋氨酸血症(小编注:高蛋氨酸血症又称高甲硫氨酸血症,是一种先天性氨基酸代谢异常疾病,因基因突变造成将甲硫氨酸转化成S-腺苷甲硫氨酸所需的甲硫氨酸腺苷基转移功能缺乏,造成甲硫氨酸堆积而导致血液中甲硫氨酸的浓度升高)和肝脏慢性SAMe缺乏。MAT1A-KO小鼠在8月龄时发生大泡性脂肪变性(小编注:大泡性脂肪变性是指细胞内出现大脂滴的脂肪变性。当肝脏内甘油三酯含量超过肝湿重的5%~10%时,许多肝细胞相继出现脂滴,随着脂肪含量的增加,各个肝细胞内的小脂滴可融合成直径大于25㎛的单个大脂滴,细胞核和细胞器被挤压移位至脂滴边缘,但细胞非脂肪部分的容积通常无显著变化),在18月龄时一半的MAT1A-KO小鼠会发展为肝细胞癌。
研究人员观察到MAT1A-KO幼崽的死亡率增加,只有54.8%的幼崽存活到断奶,而WT幼崽断奶后存活率为76.8%(图2A)。此外,研究人员还发现MAT1A-KO小鼠在21至90日龄期间体重增量显著低于WT 鼠,尽管在4个月大的雄性小鼠(图2B)和7个月大的雌性小鼠并没有观察到这种差异(图2B,辅图1C)。50-60日龄的WT和MAT1A-KO小鼠在夜间和白天的食物摄入量相似,这表明进食不足或进食模式的改变可能不是MAT1A-KO小鼠体重较低的原因(图2C)。MAT1A-KO小鼠的EE显著增强(图2D),且它们在自由进食时的活动略有下降(图2E),这表明观察到的体重增加的差异是由于EE增强而不是活动增强。
研究人员通过MRI分析脂肪量、腹股沟白色脂肪组织(ingWAT)/体重比和脂肪细胞大小,发现雄性MAT1A-KO小鼠(45-55日龄)脂肪含量较低(图2F-H)。MAT1A-KO小鼠ingWAT有明显的棕色化现象,这也由解偶联蛋白(Ucp1)mRNA和蛋白水平上调所证明(图2I、J)。同时,体外实验进一步证明MAT1A-KO小鼠的ingWAT在异丙肾上腺素刺激下甘油和游离脂肪酸的分泌增加,脂解反应增强(图2K)。
WT和MAT1A-KO小鼠的BAT有着相似的重量,但是通过HE染色可以看到MAT1A-KO小鼠多室脂滴大小明显减小(图2L),且Ucp1 mRNA和蛋白水平升高(图2M、N),在室温或急性冷暴露(4℃)时肩胛骨处BAT表面温度显著升高,表明产热增强(图2O、辅图1D)。
随后,研究人员观察到MAT1A-KO小鼠肝脏中不完全和完全脂肪酸氧化均增加(图2P)。检测到血清转氨酶水平未发生变化(图2Q),表明WT 和MAT1A-KO小鼠均没有肝脏损伤。接着,科研人员探究WT 和MAT1A-KO小鼠对葡萄糖代谢的影响,结果发现MAT1A-KO小鼠葡萄糖耐量增强,丙酮酸耐量增强,胰岛素敏感性差异不明显(辅图1E)。
以上研究结果表明,由于缺乏MAT1A引起的肝脏SAMe的慢性缺乏,即使在喂养条件下也会诱导高分解代谢状态,最终导致出生后死亡率的增加。这种分解代谢反应与WT小鼠禁食后观察到的相似,同样以SAMe水平下降为特征,表明SAMe水平的持续下降可以作为限制性进食的代谢传感器来诱导分解代谢反应。
拓展阅读
脂肪酸β-氧化速率的测定
液氮冷冻组织首先在缓冲液(25 mM Tris-HCl,500 nM 蔗糖溶液,1 mM EDTA-Na2,pH 7.4)中使用匀浆器匀浆。然后将组织匀浆后超声处理10s,离心(420 X g,10min,4℃)。对上清液进行蛋白质测定,每次测定总蛋白500 ug,最终体积为60ul。反应开始时,将样品与340ul含有0.2 mM 0.5 uCi/ml[1-14C]-棕榈酸于37℃下在覆盖有Whatman纸盖的Eppendorf管中孵育30分钟。通过注入200ul 3 M高氯酸停止反应,并将Whatman纸帽中浸渍1 M NaOH以捕获释放的二氧化碳。在室温下1h后,取出滤纸,并对酸化介质进行离心(21000×g,10min,4℃)以去除颗粒物。然后借助闪烁计数器测量捕获的二氧化碳的放射性(相当于完全氧化)和上清液中酸溶代谢产物(ASM)的放射性(相当于不完全氧化) 。
拓展阅读
脂肪酸的完全氧化和不完全氧化
脂肪酸的β-氧化,首先脂肪酸由脂酰-CoA合成酶激活为脂酰-CoA,脂酰-CoA由脂酰-CoA脱氢酶反应为反烯酰-CoA,反烯酰-CoA由烯酰-CoA合成酶合成为β-羟酰CoA,β-羟酰-CoA由羟酰-CoA脱氢酶反应为β-酮酰CoA,最后β-酮酰CoA由硫酯酶分解成少了两个碳的脂酰-CoA和一分子乙酰-CoA,乙酰-CoA可以进入三羧酸循环继续被彻底氧化(图a)。在饥饿或者病理状态下,为了维持体内血糖稳定,三羧酸循环中的草酰乙酸会作为糖异生的原料被消耗,肝细胞中的三羧酸循环受到影响,导致脂肪酸β-氧化得到的乙酰-CoA大量堆积而不能完全氧化,产生了不完全氧化的情况生成酮体(图b)。脂酰-CoA由乙酰乙酯-CoA硫解酶反应为乙酰乙酯-CoA,随后乙酰乙酯-CoA和乙酰-CoA由HMG-CoA合成酶合成为β-羟甲基戊二酸,之后被HMG-CoA裂合酶分解为乙酰乙酸,乙酰乙酸可以通过乙酸脱羧酶、脱氢酶自发变成丙酮和β-羟丁酸。
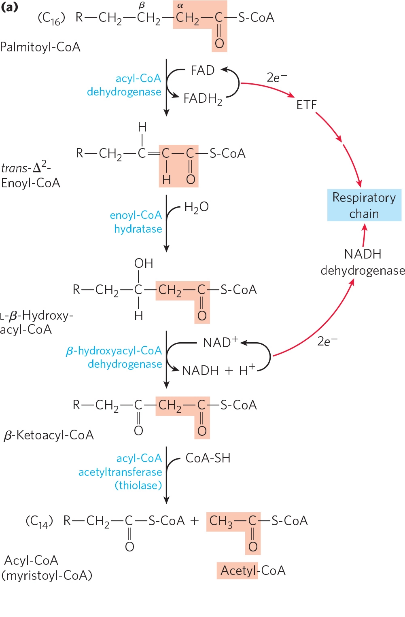
图a 脂肪酸完全氧化

图b 脂肪酸不完全氧化
拓展阅读
蛋氨酸在体内的代谢途径
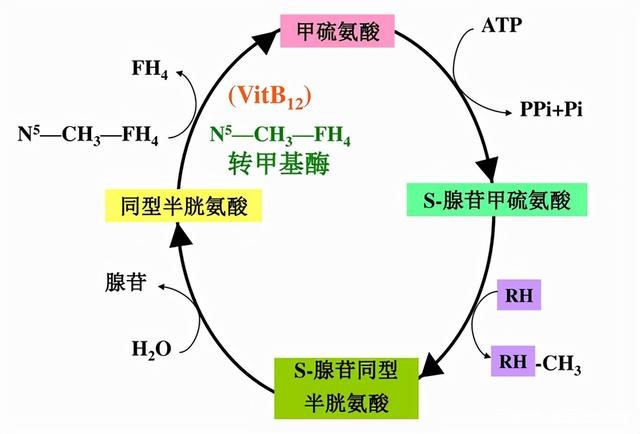
蛋氨酸(methionine,Met,又称甲硫氨酸)无法在动物机体内自身生成,必须由外部提供的必需氨基酸。Met在各种动物机体、各种组织细胞中参与代谢活动,其代谢过程是一个有多种酶参与的、复杂的生物化学反应体系。作为底物的Met参与蛋白质的生物合成,在原核生物中甲酰化的Met还是其蛋白质生物合成的起始氨基酸。Met另外还可被ATP活化转变成SAM,作为甲基供体参与肌酸、胆碱的合成;同时在代谢中还有转硫作用。甲硫氨酸循环(methionine cycle)是Met(甲硫氨酸)在体内最主要的分解代谢途径,其能够通过转甲基作用而提供甲基。在转甲基反应前,甲硫氨酸必须在腺苷转移酶的催化下与ATP反应,生成S-腺苷甲硫氨酸(SAM),SAM中的甲基成为活性甲基,SAM又称为活性甲硫氨酸。SAM经甲基转移酶催化,将甲基转移至另一种物质,使其甲基化,而SAM去甲基后生成S-腺苷同型半胱氨酸(即S-腺苷-L-高半胱氨酸(SAH)),后者脱去腺苷生成同型半胱氨酸。同型半胱氨酸再接受N5-CH3-FH4上的甲基,重新生成甲硫氨酸,形成一个循环过程,称为甲硫氨酸循环。它的生理意义主要是由N5-CH3-FH4供给甲基生成甲硫氨酸,再通过此循环的SAM提供甲基,以进行体内广泛的甲基化反应,因此N5-CH3-FH4可看成是体内甲基的间接供体。
参考文献
[1] Brosnan J T,et al.Journal of Nutrition,2006,136( 6) : 1636S-1640S.
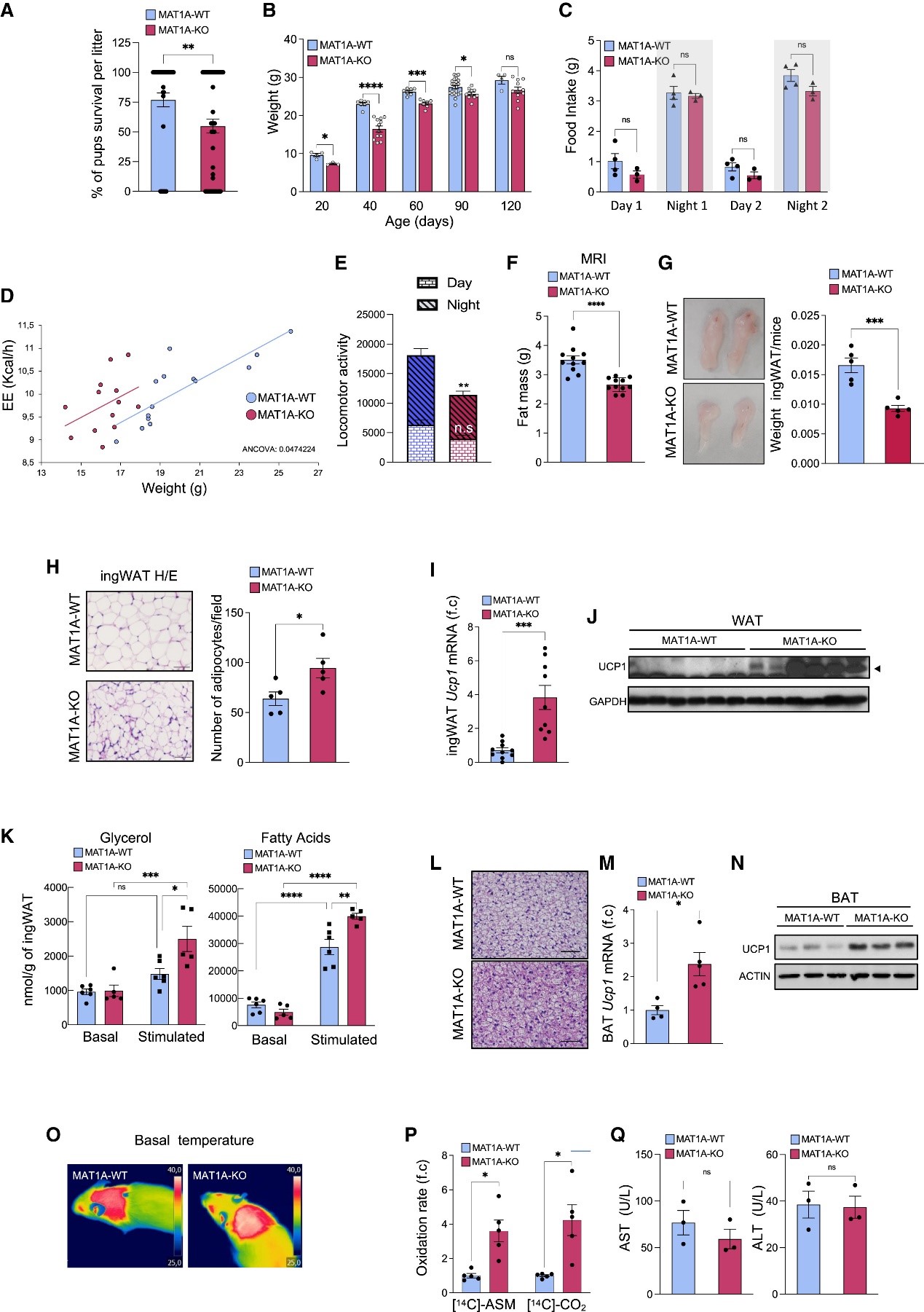
图2 MAT1A-KO小鼠体重增加减缓、能量消耗和出生后死亡率增加
3.MAT1A-KO小鼠的在禁食期间体重减轻,诱导肝脂肪酸β氧化、ATP生成、内质网应激和禁食期间肝损伤
接下来,研究人员进一步研究了MAT1A缺乏的小鼠模型,并检查它们对禁食的反应。为此,研究人员使用50-60日龄(脂肪变性出现之前)的MAT1A-KO小鼠,并对其进行36小时的饥饿处理。结果发现MAT1A-KO小鼠在禁食期间比MAT1A-WT小鼠表现出更明显的体重减轻(图3A、辅图2A)。
通过MRI分析脂肪量和ingWAT的重量可以看出,禁食导致MAT1A-KO小鼠的脂肪含量降低(图3B,C,辅图2B)。同时,体外实验进一步证明禁食导致MAT1A-KO甘油分泌的增加,表明脂肪分解增强(图3D)。然而,研究人员没有观察到MAT1A-KO ingWAT中脂肪酸分泌的增加,这可能是由于禁食24小时后ingWAT中储存的脂质消耗过多或脂肪酸氧化增强(辅图2C)。
此外,在禁食24小时和36小时后MAT1A-KO小鼠的EE明显增强(图3E),BAT多房性脂滴变小(图3F)和UCP1蛋白水平升高(图3G)。重要的是,如作者团队之前的研究所证实的那样,在WT小鼠的BAT和ingWAT中无法检测到MAT1A(图3G、辅图2D),而MAT2A在来自MAT1A-KO小鼠的BAT中没有差异表达(辅图2E),这表明在MAT1A-KO小鼠脂肪组织中观察到的影响可能是由于肝脏MAT1A表达缺乏引发的全身效应。
虽然研究人员没有观察到在喂食或禁食36小时的WT和MAT1A-KO小鼠的BAT和ingWAT中脂肪生成基因Fas、Srebp以及脂肪分解基因Hsl和Mgl的表达有显著差异,但他们发现禁食36小时后,MAT1A-KO小鼠的ingWAT中Atgl mRNA的表达显著增加(辅图2F),这与在这些组织中观察到的脂肪分解增强一致(图3D)。其次研究人员没有发现WT和MAT1A-KO小鼠之间血清胰高血糖素水平的差异,这表明在MAT1A-KO小鼠中观察到的影响可能不是由于胰高血糖素对禁食反应的作用增加所致(辅图2G)。
MAT1A-KO小鼠在禁食条件下的肝脏显示ASM氧化(代表不完全脂肪酸棕榈酸氧化)显著增加(图3H),这与血清酮体水平升高和血清甘油三酯(TGs)水平降低相关(辅图2H、I)。重要的是,研究人员发现从禁食的MAT1A-KO小鼠分离的肝细胞在基础和刺激条件下(胰高血糖素治疗)产生更多的ATP(图3I)。
接下来,研究人员评估了SAMe对禁食期间MAT1A-KO肝细胞耗氧量(OCR)、ATP生成和β氧化的影响。从禁食24小时的MAT1A-KO小鼠中分离的肝细胞显示出增强的基础呼吸、ATP相关的呼吸、最大呼吸和非线粒体耗氧量,而在MAT1A-KO肝细胞中添加SAMe会消除这种影响(图3J、辅图2J)。SAMe也阻止了MAT1A-KO肝细胞中脂肪酸氧化活性的增强(图3K)。总之,这些结果表明,在MAT1A-KO小鼠中,较低的肝脏SAMe水平增加了禁食期间的肝细胞呼吸、β氧化和ATP的产生。
有趣的是,研究人员观察到禁食24和36小时会导致MAT1A-KO小鼠的肝损伤,即血清丙氨酸转氨酶(ALT)水平升高,p38MAPK、cJun的磷酸化增强,以及真核起始因子-2ɑ(eIF2ɑ)和肌醇需求酶1(IRE1)的磷酸化增加,表明内质网应激(ERS)增加,发生肝损伤(图3M-L,辅图3A)。同样,MAT1A-KO小鼠的肝脏在经过5天60% CR后cJun和IRE1磷酸化增强(辅图3B)。
SAMe参与GSH生成,已知GSH在肝脏中起保护和抗氧化作用。为了研究GSH合成减少是否与禁食MAT1A-KO小鼠观察到的ERS增加有关,研究人员用GSH前体N-乙酰半胱氨酸(NAC)治疗了禁食36小时的MAT1A-KO小鼠,结果发现NAC治疗不能阻止肝脏eIF2ɑ磷酸化。NAC治疗禁食24小时的MAT1A-KO小鼠的原代肝细胞也获得了类似的结果(辅图3C)。总之以上数据表明,谷胱甘肽水平的降低可能不是在禁食的MAT1A-KO小鼠中观察到的ERS的原因。
哺乳动物雷帕霉素靶蛋白(mTOR)调节细胞生长并协调对氨基酸可用性的反应。在HEK293T细胞中进行的实验表明,Met饥饿可以通过降低SAMe水平和促进SAMTOR与GATOR1的关联,以SAMTOR依赖的方式抑制mTORC1信号传导。因此,研究人员决定研究MAT1A-KO小鼠肝脏SAMe水平的降低是否也可以抑制mTORC1信号传导。首先研究人员没有观察到50日龄WT和MAT1A-KO小鼠在喂养或禁食36小时条件下的mTOR磷酸化有任何变化(辅图3D)。这些观察结果与之前的数据一致,表明mTOR和mTOR底物起始因子4E结合蛋白(4EBP)的磷酸化被肝脏中SAMe水平的增加或SAM治疗后强烈抑制。总之,这些数据表明mTOR信号的改变可能不是在MAT1A-KO小鼠中观察到的影响的原因。
接下来,研究人员探究了MAT1A-KO小鼠在禁食期间和5天内60% CR观察到的肝损伤和ERS的增加是否仅仅是适应过程中应激的结果,或者它们是否可能与更长时间的CR方案相关。结果发现,用ADF或40% CR喂养2个月的MAT1A-KO小鼠,ERS也显著增加(图3N、O)。同样,在连续30个24小时的ADF期间,MAT1A-KO的体重减轻百分比与WT小鼠相比也不断提高。而在初始阶段,40% CR的MAT1A-KO小鼠也有着更高的体重减轻直到达到平台期(辅图3E、F)。值得注意的是,用ADF喂养的7只MAT1A-KO小鼠中有2只出现了肝损伤,研究人员观察到在40% CR下MAT1A-KO小鼠的ALT在2个月内显著增加(辅图3G、H)。
总之,以上数据表明,禁食期间肝脏敲除MAT1A会过度激活脂肪的使用和脂肪组织的动员,以及肝脏β氧化和ATP的产生,从而导致ERS和肝脏损伤。这表明MAT1A水平的上调在食物限制期间起保护作用,以平衡分解代谢反应。
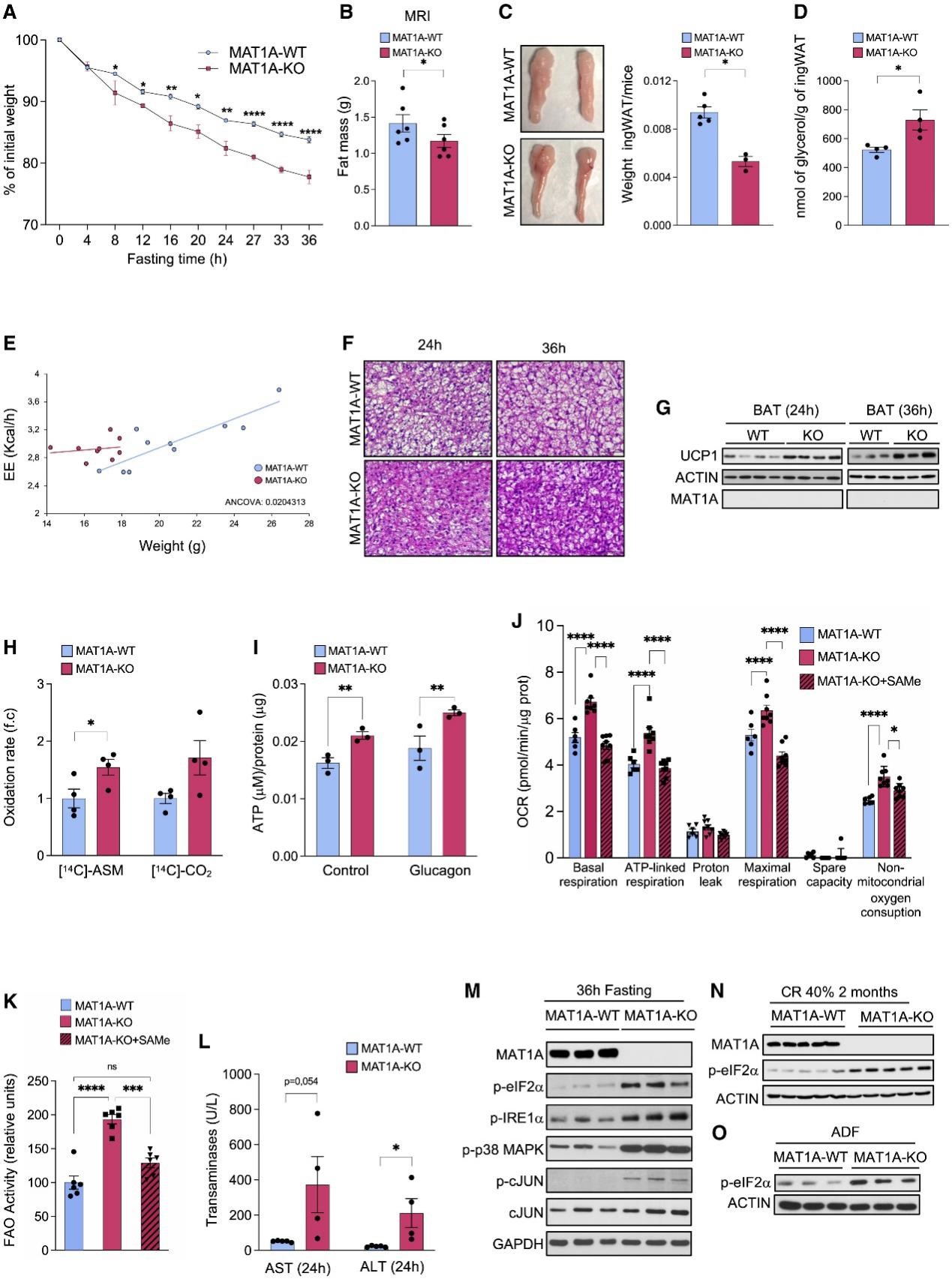
图3 禁食期间慢性MAT1A肝缺失加剧体重减轻并诱导肝损伤

辅图2 MAT1A-KO小鼠在禁食下的表型特征
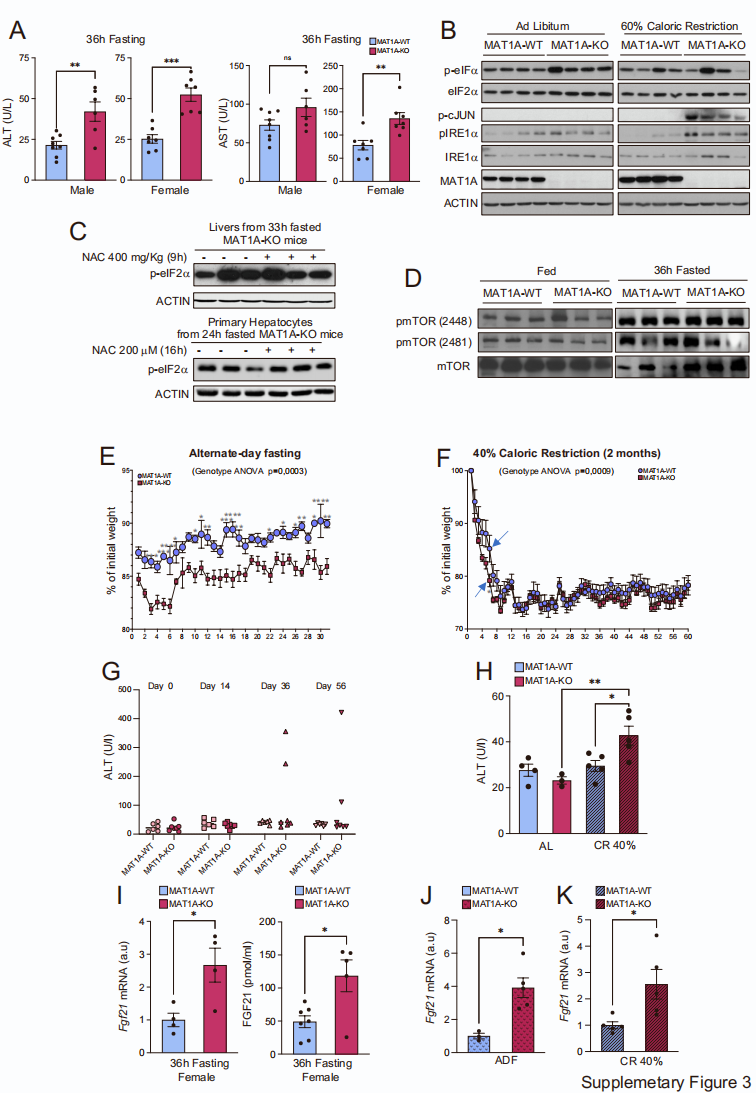
辅图3 禁食、ADF或CR对MAT1A-KO小鼠的的影响
4.MAT1A-KO小鼠中肝脏SAMe水平降低导致了肝脏中FGF21启动子低甲基化,增加了FGF21循环水平和EE
FGF21主要表达于肝脏,参与饥饿和蛋白质限制的代谢反应,在诱导肝脏脂肪氧化、生酮和糖异生、脂肪分解和WAT棕色化以及改善葡萄糖耐量方面发挥核心作用。研究人员观察到,喂食和禁食的MAT1A-KO小鼠肝脏Fgf21 mRNA水平和血清FGF21水平显著升高(图4A、B、辅图3I)。饲喂两个月的40% CR或者ADF后,MAT1A-KO小鼠肝脏中Fgf21 mRNA表达也增强(辅图3J、K)。
已有研究表明,位于Fgf21转录起始位点(TSS)下游(#10 - #21)的CpG位点的DNA甲基化率降低与肝脏Fgf21表达的增强有关。研究人员发现喂食和禁食的MAT1A-KO小鼠肝脏中CpG位点(#10 - #15)的DNA甲基化水平显著降低(图4C、D)。接下来,为了研究FGF21上调在MAT1A-KO小鼠禁食反应中的重要性,研究人员在MAT1A-KO小鼠体内沉默肝脏FGF21,有效降低了血清中升高的FGF21水平(图4E),并防止了MAT1A-KO小鼠禁食期间体脂、ingWAT重量、脂肪细胞大小的减少,以及EE的增加(图4F-I)。值得注意的是,研究人员没有在这些小鼠中观察到任何肝脏表型,包括MAT1A-KO增强的肝脏β氧化率、ERS或转氨酶升高(图4J-L)。
总之,以上研究结果表明,肝脏MAT1A的缺失诱导FGF21启动子低甲基化,可能导致肝脏FGF21合成和分泌增加,进而影响EE。另一方面,FGF21水平的增加可能不是导致限制进食后MAT1A-KO小鼠中观察到的肝损伤及β氧化增加的原因。
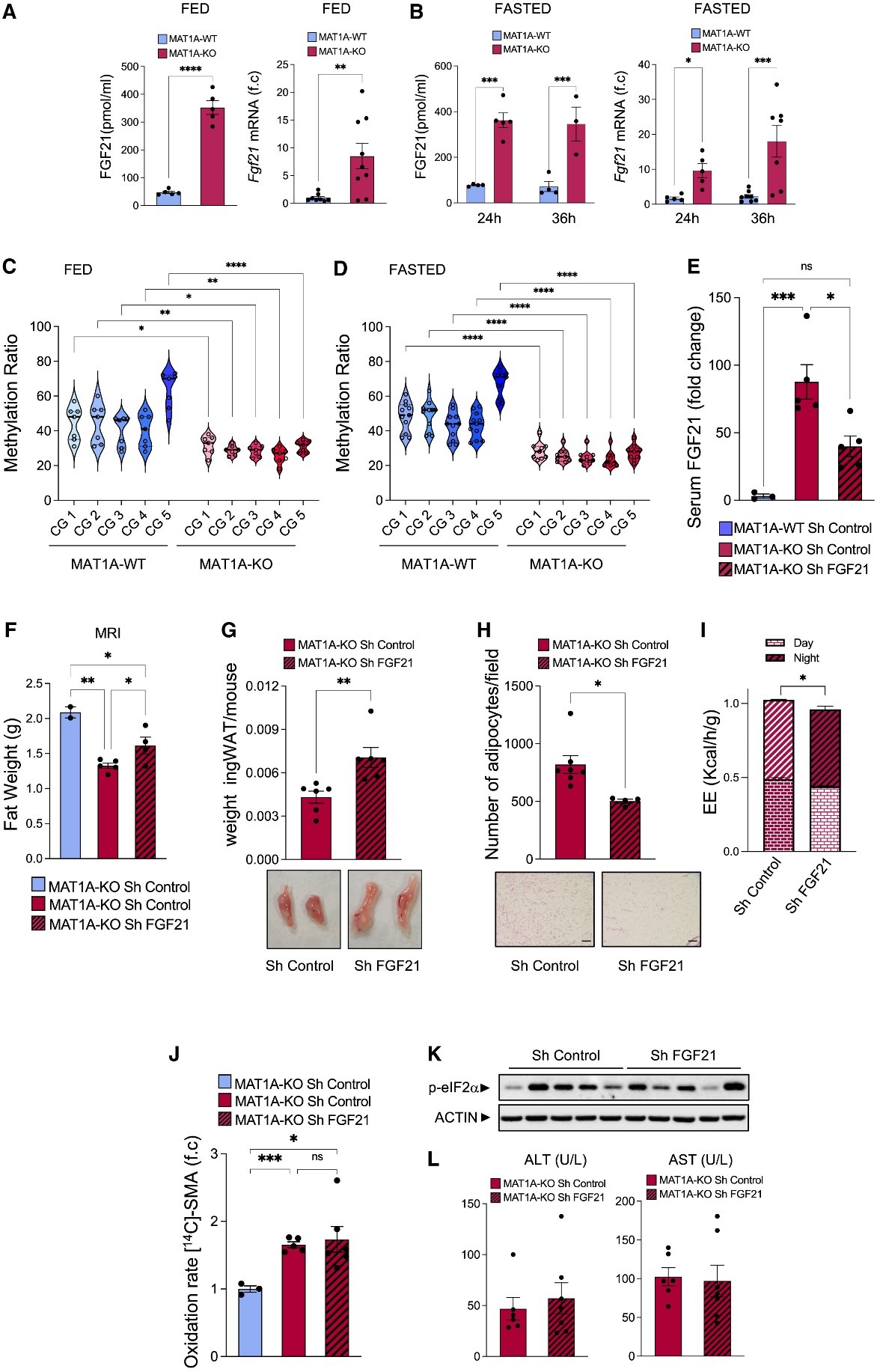
图4 MAT1A KO小鼠FGF21启动子甲基化减少增加了FGF21转录和能量消耗
5.在禁食期间的肝脏中,SAMe通过维持PEMT活性,来抑制肝脏β氧化,内质网应激和肝损伤
研究人员使用核磁共振(NMR)光谱进行了代谢通量组学实验,以跟踪13C标记的Met参与其他细胞代谢物的情况。为此,在小鼠处死前30分钟,研究人员在禁食24小时的WT和MAT1A-KO小鼠中静脉注射13C标记的Met。结果发现,在肝脏裂解物中Met循环的几种代谢物(包括SAMe、SAH、PC和同型丝氨酸)以及糖异生的代谢物(包括3-磷酸甘油、果糖-1,6-二磷酸、葡萄糖-6-磷酸和葡萄糖)和乳酸中都有放射性标记。值得注意的是,研究人员发现MAT1A-KO小鼠的SAMe、同型半胱氨酸、同型丝氨酸、乳酸和果糖1,6二磷酸代谢物中13C标记物的数量减少,Met、3-磷酸甘油、葡萄糖-6-磷酸和葡萄糖中13C标记物的数量增加(图5A、B),这表明禁食期间MAT1A的缺失导致Met的代谢通量从SAMe合成转向葡萄糖合成。这种移位可能是由于转甲基化和转硫化途径的减少(由于缺乏MAT1A)和Met转氨化途径的增加,这在MAT缺乏症的成年人中很常见。
有趣的是,在30分钟的标记过程中,也发现13C标记掺入PC的趋势有所减少。为了证实这一点,研究人员通过基于31P-NMR的磷酸化组学测量了总PC和PE含量,发现24小时饥饿的MAT1A-KO小鼠肝脏中PC和PC/PE比率明显下降(图5C)。PC合成的两种途径之一是通过肝脏PEMT对PE进行三甲基化,这一反应需要三分子SAMe。PEMT的缺失导致PC/PE比率的大幅降低,从而导致线粒体呼吸和ATP产生的增加。之前的研究表明,自由喂养的MAT1A-KO小鼠中SAMe利用率的降低会导致PEMT活性和PC/PE比率的降低。这些数据表明,在禁食的MAT1A-KO小鼠中观察到的β氧化、ATP合成、ERS和肝损伤的增强可能是由于PEMT活性的降低。为了证明这一点,研究人员在禁食36小时之前对肝脏中的PEMT进行了沉默,并发现这些小鼠的肝脏Pemt基因表达明显降低(图5D),导致肝脏PC含量和PC/PE比值降低(图5E)。PEMT沉默并没有导致更明显的体重降低(图5F)和ingWAT重量(图5G)或FGF21基因或蛋白表达的增强(图5H)。相反,禁食期间Pemt沉默诱导肝脏β氧化,如棕榈酸完全氧化(图5I),并伴有ERS(如eIF2ɑ磷酸化增强)和AST的增加(图5J、K)。在Mat1a沉默后,也观察到由Pemt沉默诱导的一些效应,类似MAT1A-KO小鼠中的数据(辅图4A-D)。最后,正如预期的那样,禁食期间FGF21沉默并没有导致MAT1A-KO肝脏中PC含量下降的恢复(辅图4E),这表明在限制进食方案中,MAT1A-KO小鼠肝脏中FGF21水平的升高可能不参与调节MAT1A-KO小鼠的PEMT功能。
总之,以上结果表明在禁食期间,SAMe水平可能对调节PEMT在肝脏β氧化、内质网应激和未折叠蛋白反应(UPR)中的功能至关重要。因此,禁食期间SAMe合成的过度减少可能导致内质网PC/PE比率的降低,诱发内质网应激,最终导致肝损伤。
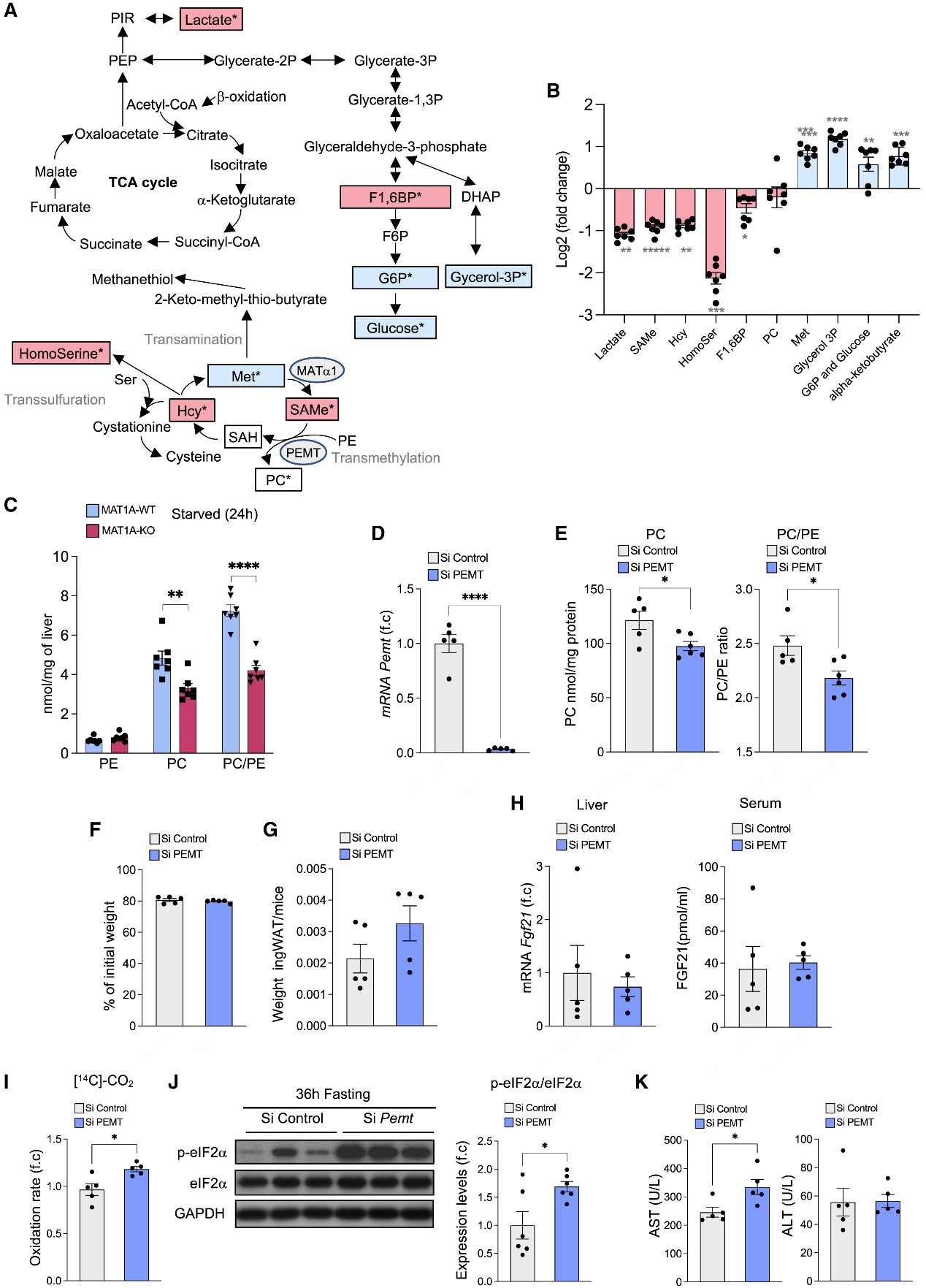
图5 禁食期间Met循环通量对SAMe和PC合成的影响
辅图4 禁食期间肝脏需要MAT1A来调节β氧化、PC合成和ERS
6.肝脏低SAMe水平增强线粒体与内质网的相互作用
线粒体的β氧化是由内质网向线粒体释放Ca2+推动的。在禁食期间,内质网的某些区域可逆地与被称为MAM的接触点的线粒体捆绑在一起,以利于Ca2+流动。喂食后,葡萄糖减少小鼠肝脏中MAMs的数量,损害线粒体呼吸。这些观察结果使研究人员假设肝脏MAT1A的缺失,进而降低肝脏SAMe水平的降低,并调节线粒体与内质网的相互作用。因此,研究人员使用透射电子显微镜(TEM)检查了喂养和禁食24小时的WT和MAT1A-KO小鼠的肝脏切片,并测量了线粒体和内质网之间的接触程度。和预期一样,在WT小鼠中禁食诱导线粒体和内质网之间的接触增加。重要的是,研究人员发现与进食和禁食条件下的WT小鼠相比,MAT1A-KO小鼠肝细胞线粒体和ER之间的接触百分比显著增强(图6A、B)。这些数据表明,肝脏SAMe水平的降低有利于ER-线粒体接、在触点的形成,这可能促进Ca2+向线粒体转移和线粒体呼吸。
拓展阅读
线粒体的β氧化与Ca2+
Ca2+可以通过多种方式调控肝脏线粒体的β氧化。其中一些机制包括:1)激活线粒体蛋白激酶:Ca2+可以激活线粒体蛋白激酶,这一酶促使线粒体内膜通透性转变。这种转变可能增加膜上蛋白质的磷酸化,从而促进脂肪酸的内部转运,使其更容易进入线粒体进行β氧化。2)激活脂肪酸合酶:Ca2+还可以通过激活脂肪酸合酶,促进脂肪酸的运入线粒体内,以供β氧化使用。这是因为脂肪酸需要与辅酶A结合才能进入线粒体,并且Ca2+可能有助于促进这一过程。3)活化脂肪酸氧化酶:Ca2+还可以与线粒体内的脂肪酸氧化酶相互作用,从而提高酶的催化活性,促使脂肪酸更快地进行β氧化,产生ATP。3)细胞信号转导:Ca2+可以影响其他细胞信号转导通路,如激活激酶、AMP激活蛋白激酶激活、磷酸化等,这些通路可能会影响线粒体的功能,包括β氧化。需要注意的是,Ca2+的作用通常是与其他细胞信号和调控机制相互作用的结果。在特定情况下,Ca2+可以激活或促进线粒体的β氧化,以满足增加的能量需求。然而,这一过程通常受到多种因素的综合调控,如激素、胰岛素和其他代谢调控因子。本文研究表明在禁食期间,内质网的某些区域可逆地与被称为MAM的接触点的线粒体捆绑在一起,以利于Ca2+流动,导致线粒体的β氧化增加,因此本文研究人员说明线粒体的β氧化是由内质网向线粒体释放Ca2+推动的(但是Ca2+流动与线粒体的β氧化的具体机制尚未阐明)。
参考文献
[1] Theurey P,et al.J Mol Cell Biol. 2016;8(2):129-143.
7.在禁食期间MAT1A定位于MAMs,调节局部SAMe合成
PEMT主要定位于MAMs,在饥饿期间维持PC合成和磷脂酰丝氨酸(PS)进入线粒体(图6C)。因此,禁食后MAT1A-KO小鼠中SAMe可用性的降低也会降低MAMs的PMET活性,导致PC/PE的降低(图6C)。
根据上述发现,研究人员假设在禁食期间,合成的MAT1A蛋白可以定位于靠近PEMT的ER/MAM区域,从而局部产生其活性所需的SAMe(图6C)。事实上,研究人员发现24小时禁食、ADF、急性CR(5天60%)或慢性CR(2个月40%)都导致细胞器中MAT1A蛋白的增加(图6D、E)。然后,研究人员使用差速离心亚细胞分离方法从新鲜肝组织中获得MAMs、ER、粗线粒体和纯粒线体(MP)组分。随后发现禁食6小时后,在MAMs中检测不到MAT1A蛋白,但禁食16-24小时后,MAT1A蛋白高度富集(图6F、辅图4F)。以上研究结果与Fig 1C一致,研究人员分析了禁食后Mat1a mRNA和MAT1A蛋白水平,并发现Mat1a mRNA和MAT1A蛋白的表达在0-6h 变化不明显,12小时到24小时逐渐增加 (图6G、辅图4G)。
接下来,研究人员通过测量从喂食和24小时饥饿的WT和MAT1A-KO小鼠中分离的总肝脏提取物和粗线粒体提取物(包括MAMs和线粒体)中的SAMe和SAH,研究了禁食期间MAMs处MAT1A的定位是否会导致该特定区室中SAMe合成的区室化。营养应激(禁食或CR)没有改变WT和MAT1A-KO小鼠总肝脏匀浆的SAMe水平(图6H、辅图4H-J)。然而,引人注目的是,研究人员发现在粗线粒体提取物中,24小时禁食的MAT1A-KO小鼠的SAMe水平显著降低(图6I)。研究人员还观察到饥饿的MAT1A-KO小鼠粗线粒体中SAH水平降低,这表明饥饿期间MAT1A-KO小鼠的甲基化反应减少(辅图4K)。这些数据表明,食物剥夺或限制会在需要甲基化反应的特定亚细胞位置诱导SAMe合成的区室化,这可能是一种重要的机制,以抵消由营养缺乏引起的SAMe水平的整体降低,防止过度的β氧化和ERS,从而保护肝脏免受损害。

图6 禁食期间,MAT1A定位于MAMs
总结
在这篇文章中,研究人员阐明了肝脏S-腺苷蛋氨酸(SAMe)——一种主要的甲基供体,可以作为营养代谢传感器在小鼠中调节磷脂酰乙醇胺N-甲基转移酶(PEMT)活性、内质网-线粒体接触、β-氧化和肝脏ATP产生,以及FGF21介导的脂肪分解和脂肪组织中的产热,来微调禁食条件下的分解代谢。此外,研究人员还观察到,胰高血糖素诱导了肝脏MAT1A的表达,并使其转位到线粒体相关膜。这导致该代谢物在局部产生并作为制动器来防止过度β-氧化和线粒体ATP合成,从而减轻内质网应激和肝损伤。总之,本篇文章揭示了SAMe在禁食代谢适应中的新机制,这对于理解禁食对人体健康的影响,以及开发治疗肥胖、糖尿病和非酒精性脂肪肝等代谢性疾病的药物具有重要意义。
原文链接:https://www.sciencedirect.com/science/article/pii/S1550413123002619?via%3Dihub
关注微信公众号代谢学人
了解更多代谢前沿资讯

https://blog.sciencenet.cn/blog-3483272-1406534.html
上一篇:代谢学人Cell Metabolism:减肥难,反弹易,单核细胞来帮你!
下一篇:Cell Stem Cell:改写衰老宿命,让细胞“青春永驻”