博文
Aβ蛋白与阿尔茨海默症_MCE(MedChemExpress)
|
Section.01
Aβ 从哪来?
——APP 的“错误剪裁”是起点
Aβ 蛋白并非凭空出现,它的“前身”是淀粉样蛋白前体 (APP)——一种广泛存在于神经元细胞膜上的正常蛋白。
在健康人体内,APP 会通过非致病性途径被分解,维持大脑的正常生理功能。在某些情况下,APP 没有被“安全拆解”,而是走上了一条代价高昂的“歧路”——淀粉样变性途径,这便是 Aβ 蛋白的诞生之路。
这条“歧路”需要两步关键切割:
• 第一步:β-分泌酶 (BACE1) 会将全长 APP 切成两部分,分别是可溶性的 sAPPβ 和膜结合的 C99 片段 (CTFβ)。
• 第二步:γ-分泌酶复合物登场。这个复合物由 PS1/PS2、NCT、PEN2、APH1 四种蛋白组成,它会进一步切割 C99 片段,最终产生两种产物:Aβ 蛋白和 APP 胞内结构域 (AICD)[1]。
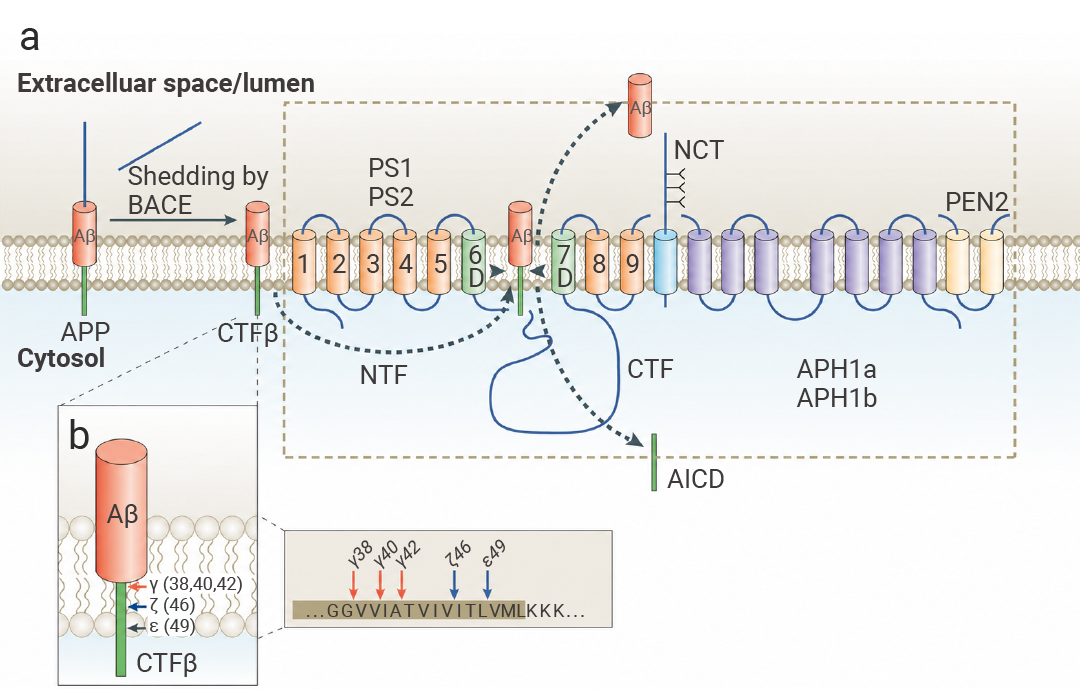
图 1. Aβ 的产生[1]。
新生成的 Aβ 蛋白单体并不安分,它们会自发“抱团”。先是组装成可溶性的 Aβ 寡聚体 (AβOs) 和原纤维,最终聚集形成不溶性的纤维沉淀,也就是我们常说的 Aβ 斑块。
值得注意的是,Aβ 单体本身毒性较弱,当前研究普遍认为,真正危险的,并不是沉积在那里的 Aβ 斑块,而是那些游走在突触之间、难以被清除的 Aβ 寡聚体和原纤维[2]。
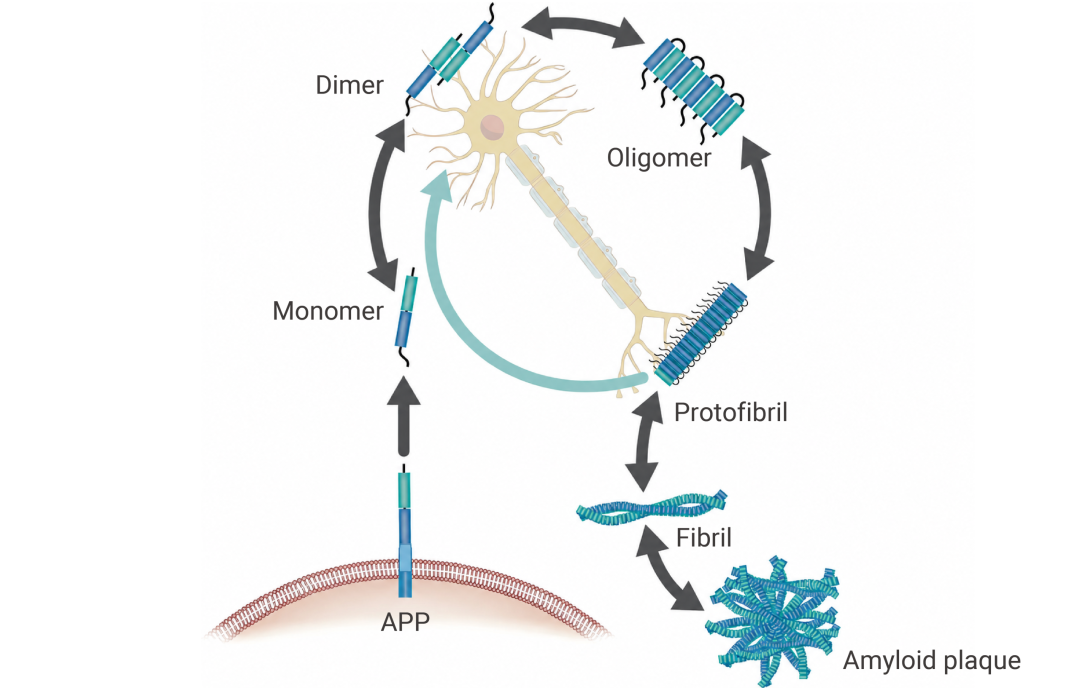
图 2. 阿尔茨海默病中的淀粉样蛋白 β 途径[2]。
Section.02
Aβ 点燃“神经炎症”导火索
——小胶质细胞的双面人生
Aβ 沉积不仅是化学问题,更是免疫警报。
一旦 Aβ 寡聚体出现,大脑的“常驻卫士”——小胶质细胞 (Microglia) 就会被激活。起初,这是好事:活化的小胶质细胞会迁移到 Aβ 沉积处,试图吞噬并清除这些“垃圾”。它们通过 TREM2、LRP1、P2X7、TLR2/4/6、CD36 等受体识别 AβO。其中,TREM2 的作用尤为关键——当 AβO 与其结合后,通过衔接蛋白 DAP12 激活 SYK 通路,促进 Aβ 降解。研究发现,TREM2 缺失会显著加重早期 Aβ 病理[3][4]。
但问题在于:这种警报如果长期拉响,守卫也会被拖垮。
这些长期暴露于 Aβ 的小胶质细胞逐渐向 DAM (Disease-associated microglia,疾病相关小胶质细胞) 状态转变,其转录组和功能特征与稳态小胶质细胞显著不同。长期处于激活状态后,小胶质细胞体积增大,却逐渐“干不动活”,吞噬能力下降,但炎症信号的释放却没有停下来。更糟的是,随着年龄增长,小胶质细胞表面的 Nogo 受体 (NgR) 表达升高,进一步抑制其吞噬功能[5]。
为了弥补清除不足,外周巨噬细胞被招募入脑,在慢性炎症背景下可能进一步放大炎症反应,形成恶性循环。
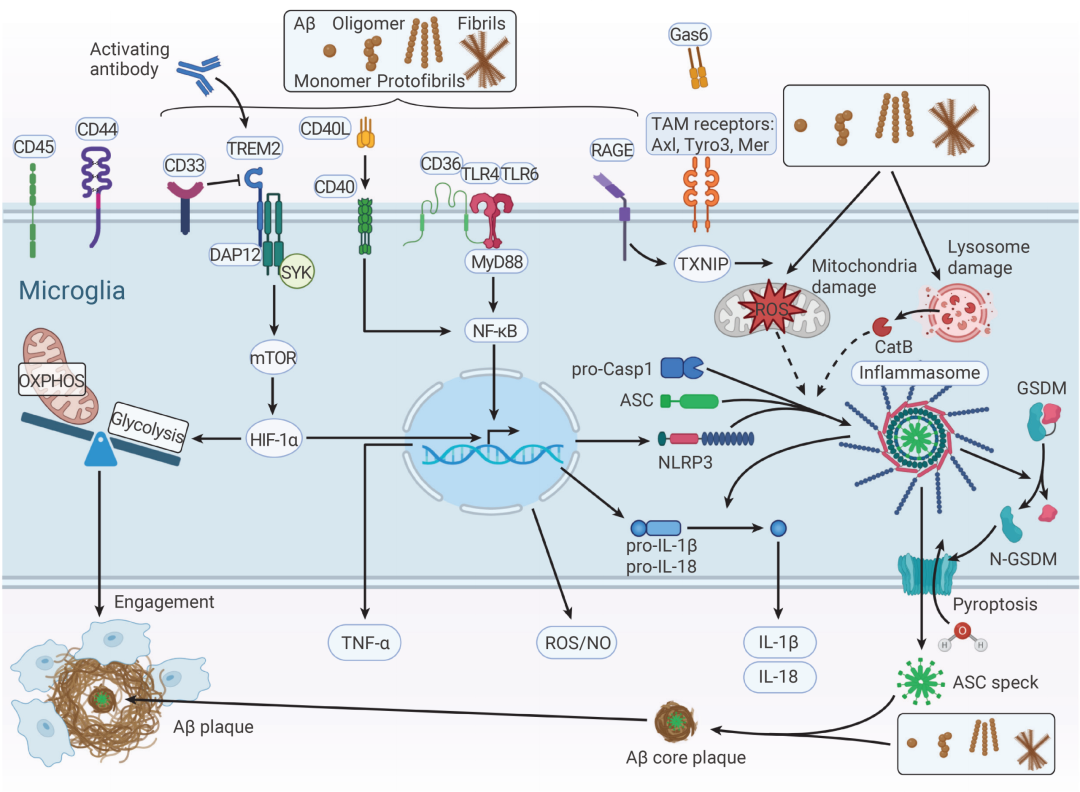
图 3. 阿尔茨海默病中的小胶质细胞激活[4]。
Section.03
星形胶质细胞
——从“支持者”变成“帮凶”
如果说小胶质细胞是前线战士,那星形胶质细胞 (Astrocytes) 原本是后勤保障——维持离子平衡、提供营养、回收谷氨酸。但在 Aβ 和炎症因子的双重打击下,它们也被“策反”了。
活化的星形胶质细胞转变为 A1 型反应性星形胶质细胞 (由小胶质细胞释放的 IL-1α、TNF、C1q 等炎症因子诱导),开始大量表达 GFAP、vimentin 等中间丝蛋白,并分泌更多炎症因子。
注意:反应性星形胶质细胞具有高度异质性,A1/A2 更多是一种功能导向的简化分类。
更危险的是,它们会上调补体分子 C3。C3 与神经元或小胶质细胞上的 C3aR 受体结合后,在神经元中导致突触丢失和认知下降;在小胶质细胞中则加剧功能障碍和炎症。
此外,星形胶质细胞释放的 APOE (APOE4 亚型风险更高) 虽能与 AβO 结合形成脂蛋白颗粒协助清除,但也可能通过 LRP1 增强小胶质细胞的炎症反应——可谓一把双刃剑。
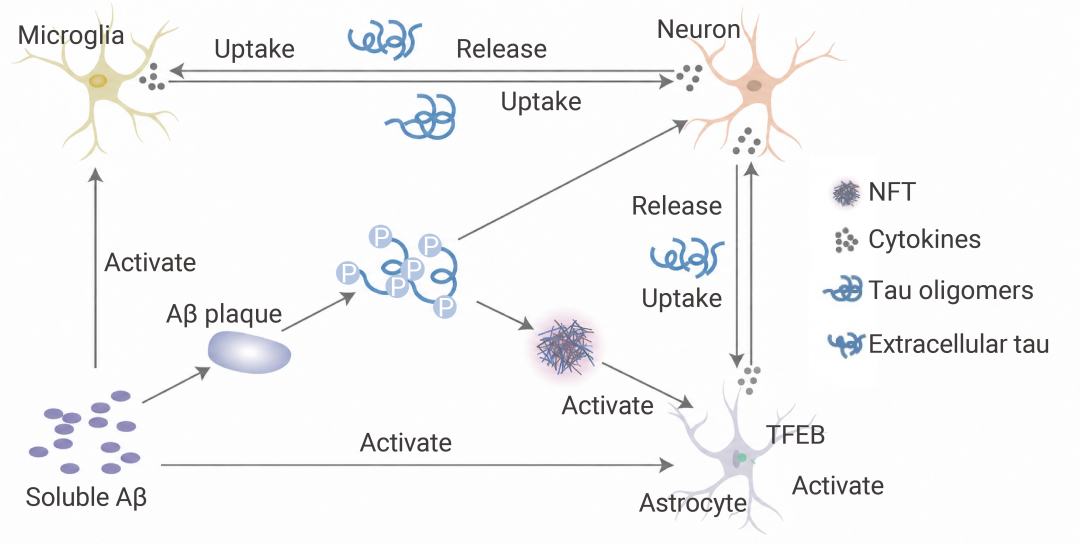
图 4. Aβ 和 tau 与小胶质细胞和星形胶质细胞的相互作用[6]。
Section.04
突触崩溃:Aβ 如何“偷走”记忆?
记忆并不是突然消失的,它是在突触一场场功能失衡中,被一点点“偷走”的。
Aβ 通过以下方式破坏突触:
• Aβ 干扰谷氨酸循环:增加释放、抑制 EAAT1/EAAT2 转运体对谷氨酸的回收,导致突触外 NMDAR 过度激活。
• 引发钙超载:过度激活的 NMDAR 引发 Ca2+ 内流,造成钙超载,一旦 Ca2+ 失控,后续反应就像被推倒的多米诺骨牌:LTP 被抑制,LTD 被放大,CDK5 和 GSK-3β 被持续激活,tau 蛋白逐步偏离正常轨道。
此外,AβO 还能结合 PrPC-mGluR5 复合物,激活 Fyn 激酶,使含 NR2B 的 NMDAR 滞留于突触膜,进一步放大钙信号。Fyn 还激活 MAPK 通路 (如 ERK、p38),同样促进 tau 病变[7]。
总之,突触功能障碍,决定了“记忆能不能形成”。
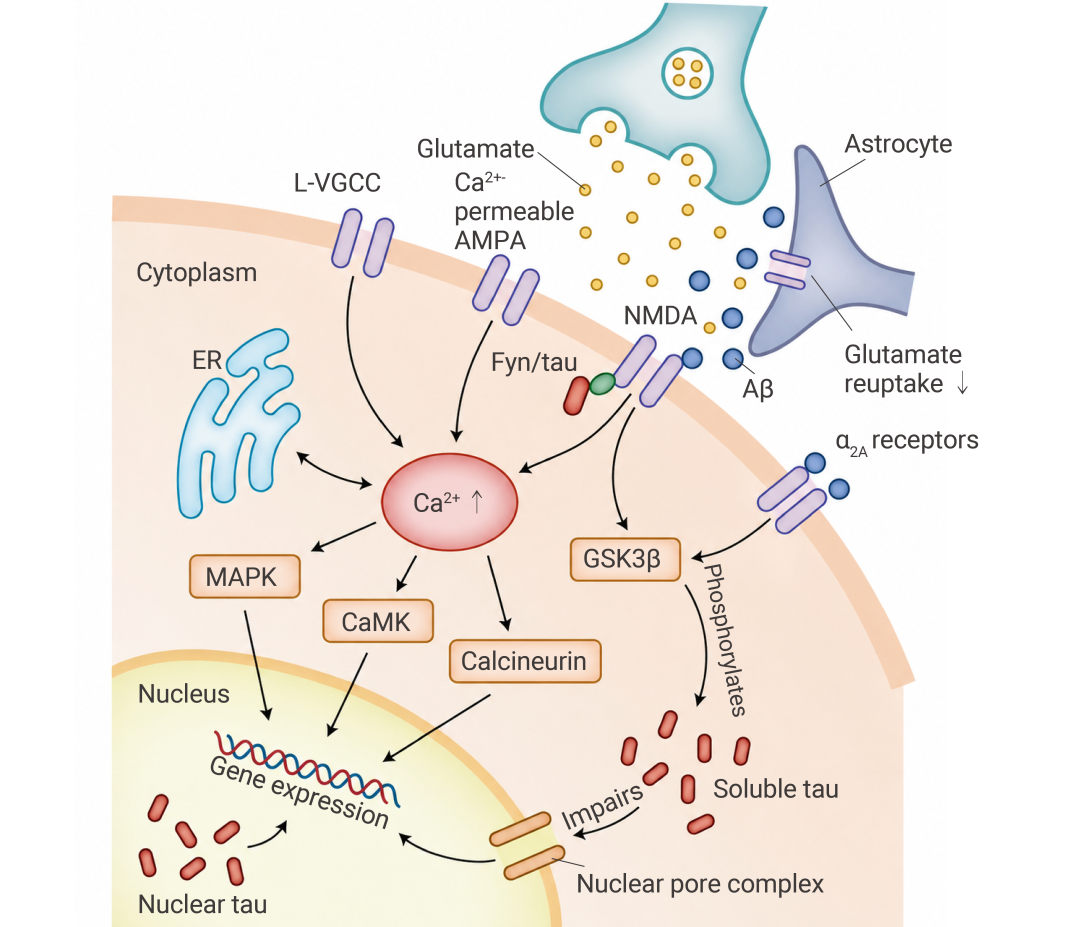
图 5. Aβ 导致神经元活动发生变化[7]。
Section.05
线粒体崩坏与 Wnt 信号中断:
雪上加霜
如果说突触功能障碍决定了“记忆能不能形成”,那么线粒体功能障碍则决定了“神经元还能不能活下去”。
Aβ 与 ABAD/HSD17B10 和 Drp1 结合,诱发线粒体过度裂变,导致 ROS 升高、ATP 合成障碍,而 tau 的过度磷酸化或 caspase-3 截短也会加剧线粒体碎片化。这间接导致 β 分泌酶和 γ 分泌酶对 APP 的淀粉样蛋白生成处理增加,同时 pTau 聚集体的积累也同步进行。
致病性 Aβ 和 pTau 会损害线粒体吞噬能力,进而导致受损线粒体增加,并引发自我传播的恶性循环。
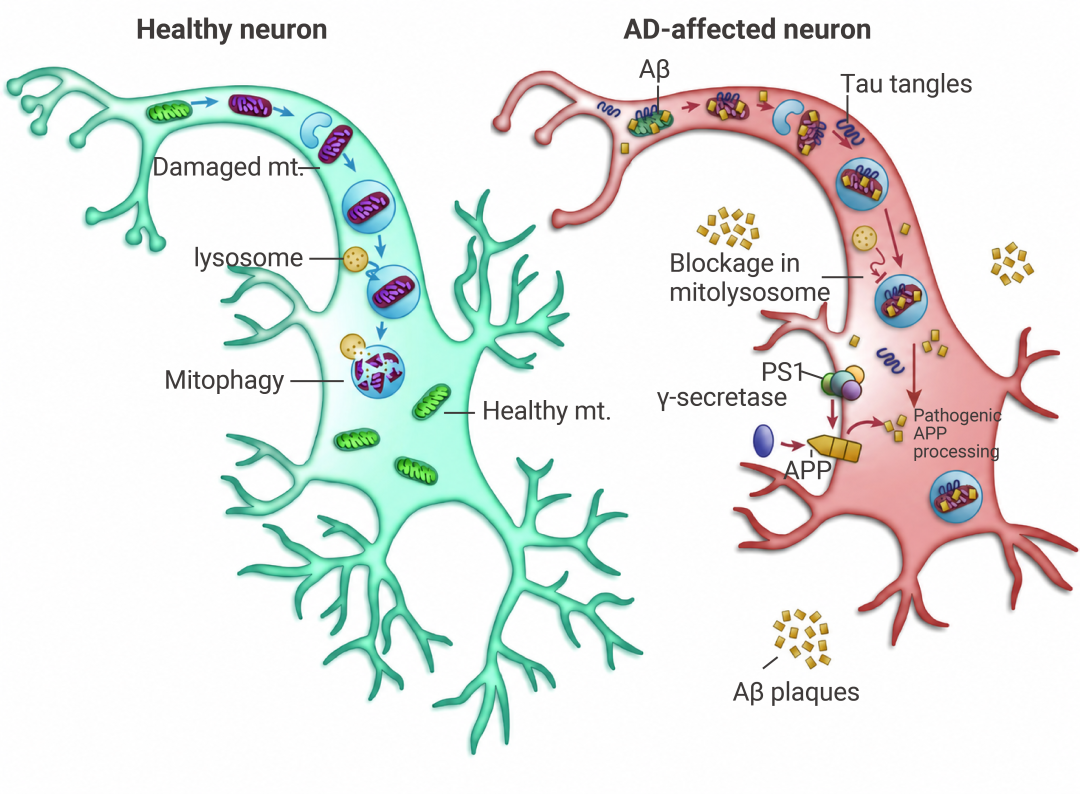
图 6. AD 中的线粒体功能障碍和线粒体吸收受损[8]。
更隐蔽的是,AβO 会促进 Wnt 拮抗剂 Dkk1 表达,竞争性阻断 Wnt 与 Frizzled 受体结合,使 β-catenin 被 GSK3β 磷酸化降解,从而抑制 Wnt/β-catenin 通路——这一通路对突触稳定和神经元存活至关重要[9]。
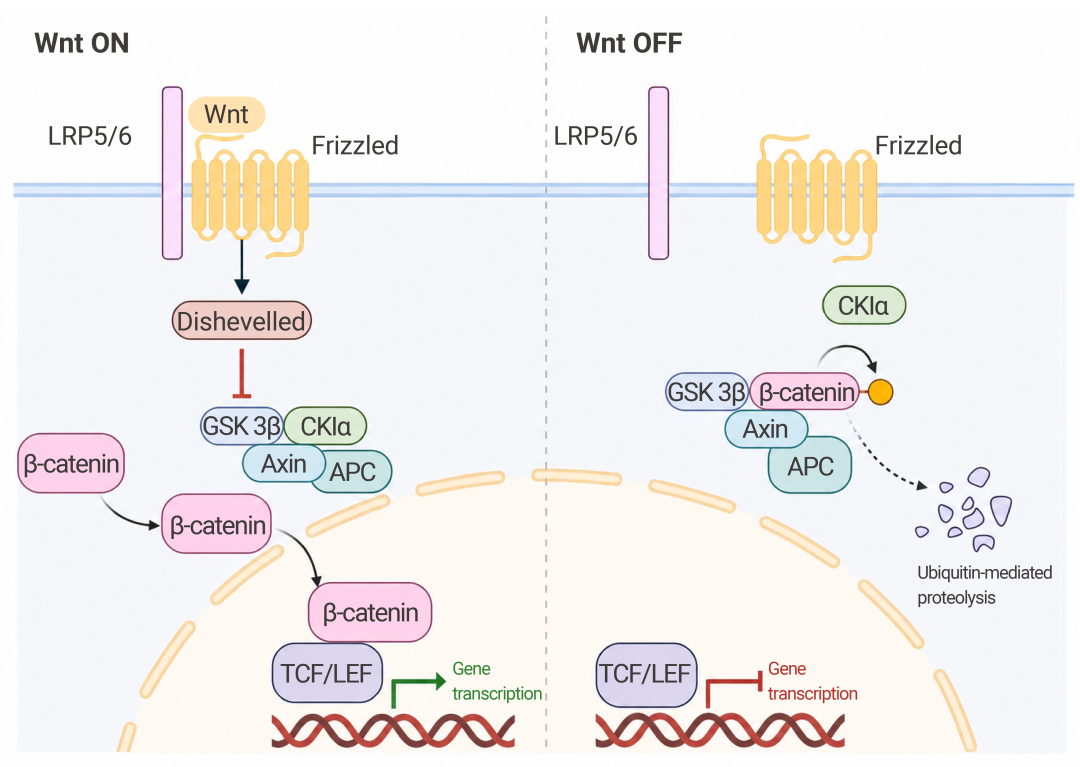
图 7. Wnt 信号通路在细胞中的作用[9]。
Section.06
希望之光:M1 受体
——潜在的“刹车”开关
面对这场多米诺骨牌式的灾难,是否还有干预窗口?
答案是肯定的。
研究发现,激活 M1 型毒蕈碱乙酰胆碱受体 (M1 mAChR) 可带来多重保护:
• 通过 PKC 和 ERK1/2 通路,促进 APP 的非淀粉样切割 (激活 α-分泌酶,抑制 BACE);
• 通过抑制 GSK3β,减少 tau 过度磷酸化;
• 缺失 M1 则会加剧 Aβ/tau 病理与认知缺陷[10]。
——这提示我们:增强胆碱能信号,或可同时阻断 Aβ 生成与 tau 病变,是极具潜力的治疗策略。
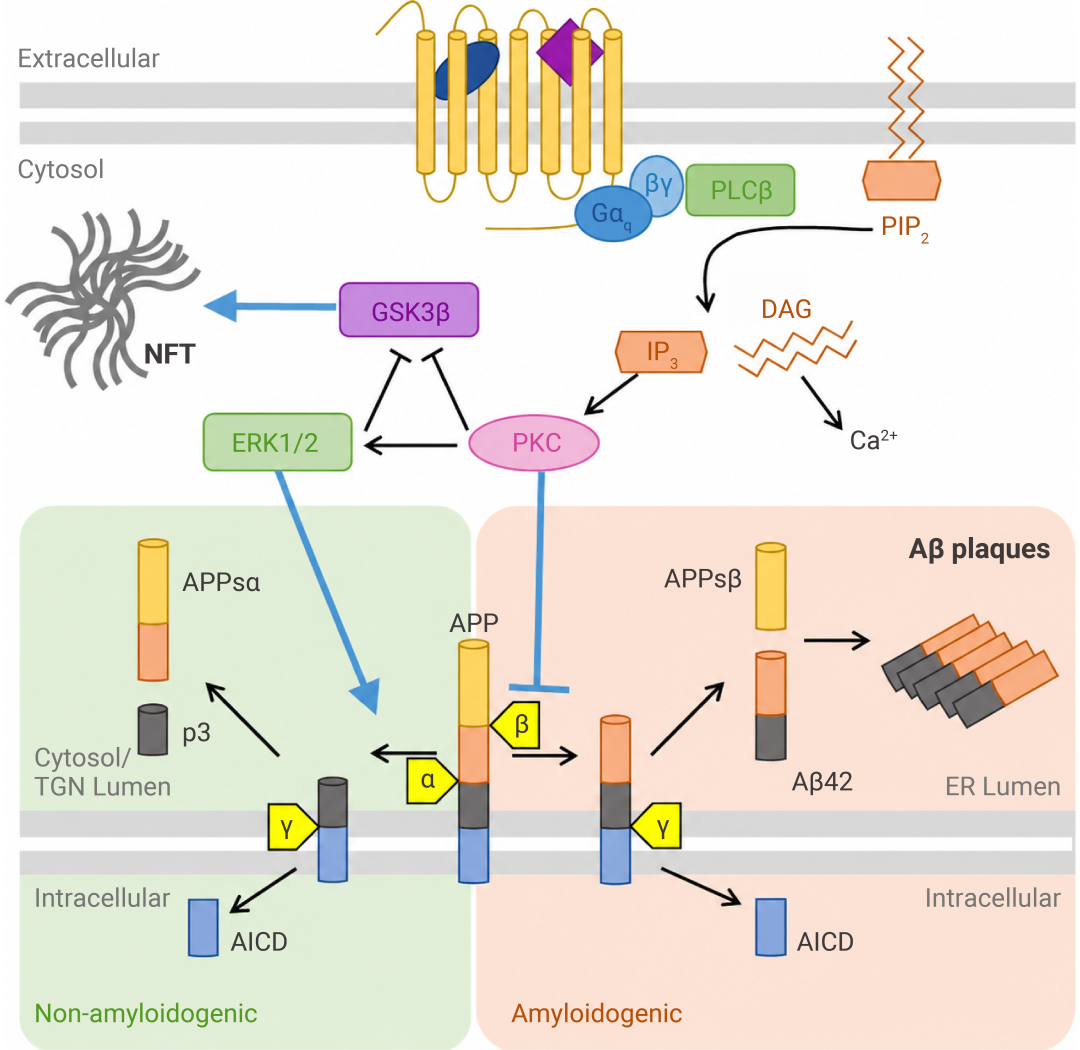
图 8. M1 mAChR 调控 AD 中淀粉样蛋白的生成过程[10]。
产品推荐
DN5355(HY-159083)
抑制Aβ和tau蛋白的聚集
Valiltramiprosate(HY-117259)
口服可用的小分子β-淀粉样蛋白β-amyloid (Aβ)抑制剂
MDR-1339(HY-14503)
口服的,透过血脑屏障的Aβ聚集抑制剂
AL002(HY-P991220)
TREM2激动剂抗体
Aducanumab(HY-P9967)
靶向聚集的β 淀粉样蛋白(Aβ) 的人单克隆抗体
Gantenerumab(HY-P99022)
特异性结合Aβ纤维和斑块
Leucomethylene blue mesylate(HY-19948)
有口服活性的二代tau蛋白聚集的抑制剂
C004019(HY-138669)
靶向tau的小分子PROTAC
CNS-11(HY-156585)
tau原纤维分解化合物
Umibecestat (HY-119689)
BACE-1抑制剂
LY2886721(HY-13240)
BACE-1抑制剂
Semagacestat (HY-10009)
y-secretase 抑制剂
DAPT(HY-13027)
口服活性的y-secretase抑制剂
lba1抗体(YA5582)(HY-P85890)
抗Iba1 的IgG1 单克隆抗体
3-Amyloid (1-42),human TFA(HY-P1363)
诱导Aβ 蛋白沉积、记忆障碍、突触功能障碍和神经发生受损
Scopolamine (HY-N0296)
诱导动物认知记忆缺陷
beta-Amyloid抗体(YA6114)(HY-P86422)
抗 Amyloid-β IgG单克隆抗体
Phospho-Tau抗体(YA3436)(HY-P83700)
抗磷酸化Tau(Ser202/Thr205) 的IgG单克隆抗体
GFAP抗体(YA755)(HY-P80139)
抗 GFAP的IgG1单克隆抗体
参考文献
[1]Haass C, et al. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007 Feb;8(2):101-12.
[2]Hampel, H., et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol Psychiatry 26, 5481–5503 (2021).
[3]Wang S, et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer's disease model. J Exp Med. 2020 Sep 7;217(9):e20200785.
[4]Mary A, et al. Immune Activation in Alzheimer Disease. Annu Rev Immunol. 2024 Jun;42(1):585-613.
[5]Wang J, et al. Nogo receptor impairs the clearance of fibril amyloid-β by microglia and accelerates Alzheimer's-like disease progression. Aging Cell. 2021 Dec;20(12):e13515.
[6]Zhang H, et al. Interaction between Aβ and Tau in the Pathogenesis of Alzheimer's Disease. Int J Biol Sci. 2021 May 27;17(9):2181-2192.
[7]Busche MA, et al. Synergy between amyloid-β and tau in Alzheimer's disease. Nat Neurosci. 2020 Oct;23(10):1183-1193.
[8]Kerr JS, et al. Mitophagy and Alzheimer's Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017 Mar;40(3):151-166.
[9]Ma J, et al. The association of the Wnt/β-catenin signaling pathway with Alzheimer's disease. Neuropharmacology. 2026 Feb 1;283:110754.
[10]Scarpa M, et al. M1 muscarinic acetylcholine receptors: A therapeutic strategy for symptomatic and disease-modifying effects in Alzheimer's disease? Adv Pharmacol. 2020;88:277-310.
https://blog.sciencenet.cn/blog-3536222-1538139.html
上一篇:细胞培养:来源、类型、培养条件、常见污染_MCE(MedChemExpress)
下一篇:药物研发:抗菌药物药效(PD)药代(PK)的给药逻辑_MCE(MedChemExpress)
