博文
肥胖的果糖生存假说  精选
精选
|
肥胖的果糖生存假说
果糖生存假说提出,肥胖和代谢紊乱可能源于对一种基于进化的生物反应(生存开关)的过度刺激,该反应旨在提前保护动物免受危机影响。这种反应的特征包括饥饿、口渴、觅食、体重增加、脂肪积累、胰岛素抵抗、全身性炎症和血压升高。这一过程是通过摄入果糖或通过多元醇途径刺激内源性果糖产生来启动的。与其他营养素不同,果糖会减少细胞内的活性能量(三磷酸腺苷),同时阻止其从脂肪储存中再生。这是通过细胞内尿酸、线粒体氧化应激、AMP激酶的抑制以及加压素的刺激介导的。线粒体氧化磷酸化受到抑制,而糖酵解得到刺激。虽然这种反应本意是适度且短暂的,但在人类中由于获得了“节俭基因”加上富含含果糖或能产生果糖的食物的西方饮食,这种反应被夸大了。我们提出,过量的果糖代谢不仅解释了肥胖,还解释了糖尿病、高血压、非酒精性脂肪肝病、与肥胖相关的癌症、血管和阿尔茨海默症痴呆,甚至老化的流行病。此外,这一假设将当前关于肥胖的假设统一起来。减少激活和/或阻断这一途径并刺激线粒体再生可能有益于健康寿命。本文是“肥胖的原因:理论、推测与证据(第一部分)”讨论会议议题的一部分。
Johnson RJ, Lanaspa MA, Sanchez-Lozada LG, Tolan D, Nakagawa T, Ishimoto T, Andres-Hernando A, Rodriguez-Iturbe B, Stenvinkel P. The fructose survival hypothesis for obesity. Philos Trans R Soc Lond B Biol Sci. 2023 Sep 11;378(1885):20220230.
1. 介绍
科学研究中经常被忽视的一个方法是调查自然及其伴随的进化力量是如何找到解决棘手问题的解决方案。这样的仿生学方法可以为基于野生动物在野外对抗逆境的巧妙方法提供新疗法的见解。然而,进化也可能无意中导致疾病。的确,有人提出,为了在资源稀缺的世界中帮助生存而进行的遗传适应(节俭基因)可能在资源丰富的世界中“适得其反”,增加了肥胖和糖尿病的风险。
在这里,我们讨论了一个最近发现的我们称之为“生存开关”的保护机制,它在资源变得稀缺之前就启动了。我们的工作表明,这种效应是由果糖介导的,与葡萄糖的主要生物功能是提供即时燃料不同,果糖的主要功能是帮助储存燃料。这些不同的生物功能是葡萄糖和果糖代谢如何调节细胞内能量水平的结果。我们讨论了果糖的不同来源以及它是如何介导其生物学作用的。我们还提出,两个事件将这一保护途径转变为导致疾病的路径。第一个是获得“节俭基因”(或更准确地说是失去了创造节俭基因型的基因),第二个事件是含有或产生果糖的食物显著增加。这两个事件导致了“生存开关”的过度激活,我们提出这正在推动肥胖和许多今天影响我们的“生命负担”非传染性疾病。
2. 果糖的来源,生存开关的触发器
果糖是一种简单糖,是水果和蜂蜜中的主要营养素。然而,在西方饮食中,它的主要来源是蔗糖(由果糖和葡萄糖结合而成)和高果糖玉米糖浆(HFCS,由果糖和葡萄糖混合而成,通常果糖浓度略高,因为测试表明人类更喜欢稍甜一点的果糖)。如今这些“添加糖”占总体能量摄入的约15%,有些群体摄入高达20%或更多。
果糖还可以从体内的葡萄糖生成(图1)。这发生在葡萄糖水平(即底物)过高时,如糖尿病、摄入高血糖指数的碳水化合物后,以及高碳水化合物饮食中。葡萄糖转化为果糖的酶促过程称为多元醇途径,其限速酶是醛糖还原酶(AR)。AR可因压力而激活,如脱水(高渗透压)、饥饿(如缺血)或缺氧。盐分食物和酒精(都会提高血清渗透压)以及果糖本身也会刺激果糖的生成。鲜味食物(富含谷氨酸和核苷酸,如一磷酸腺苷(AMP)和一磷酸肌苷(IMP))也会产生尿酸,从而刺激果糖的产生。所有这些营养素都会在肝脏中刺激果糖的生成,足以完全激活生存开关。果糖的生成也可以在其他器官中被刺激,如大脑、肾脏、血管和心脏。实际上,果糖是在大脑中局部产生的,当血糖过高时;在心脏中,当心脏缺血时;在肾脏中,当缺血再灌注损伤时;在裸鼹鼠爬行通过缺氧隧道的循环中;以及在胎盘和胎儿中,在第一个孕期期间,在胎盘循环完全建立之前的缺氧期。由于酒精能提高渗透压,它也可以刺激果糖的产生。
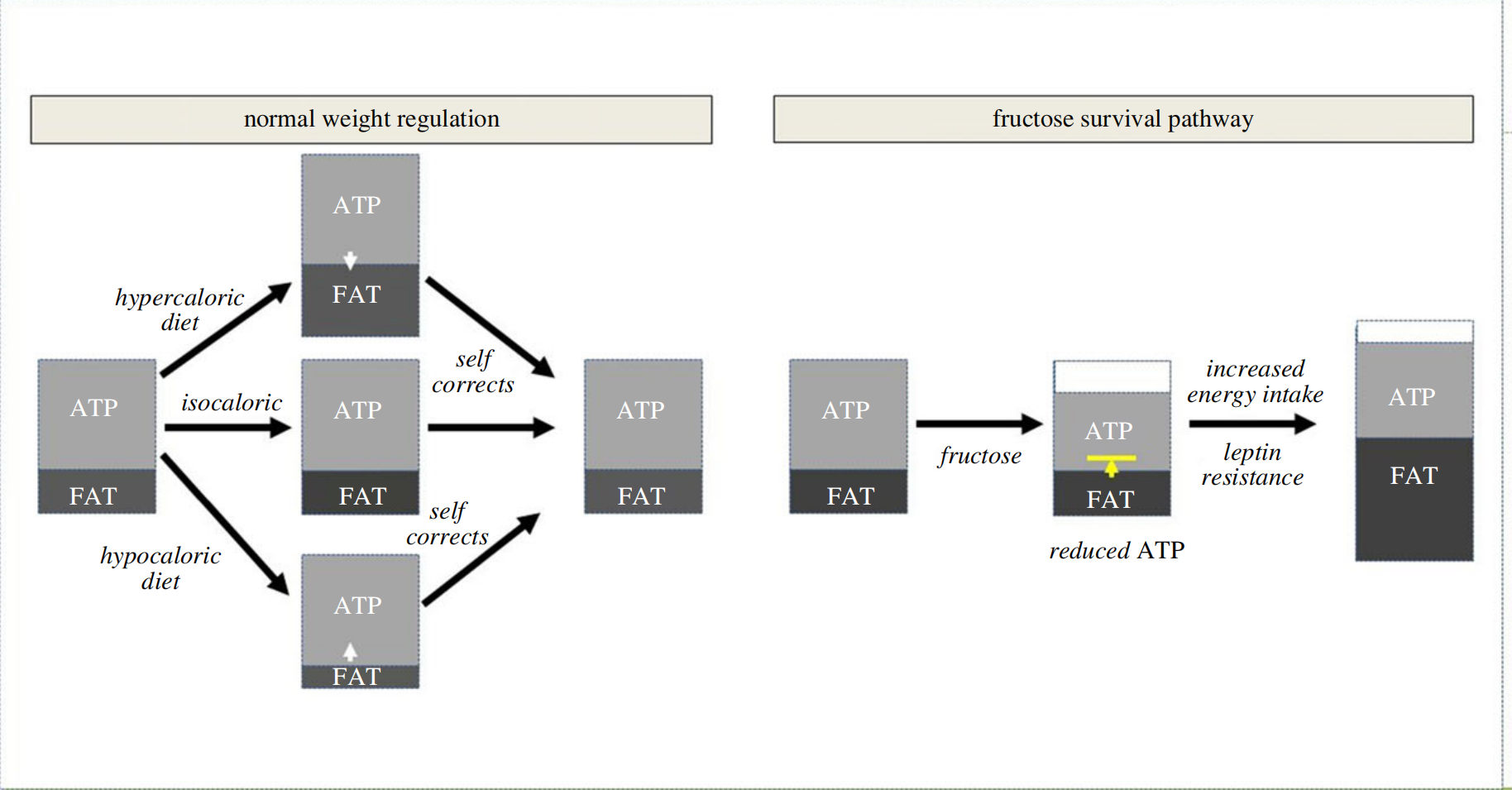
图1.果糖改变体重调节。在正常情况下,大多数动物严格调节它们的体重。如果它们被喂食高热量饮食,它们会增加体重,如果它们被给予低热量饮食,它们会减轻体重,但当它们被允许恢复正常饮食时,它们会自发地校正到正常体重。相比之下,果糖通过降低细胞内ATP的同时阻止脂肪储存中ATP的补充来起作用。经过几天到几周的时间,动物发展出瘦素抵抗,导致能量摄入增加。然而,由于氧化应激的抑制,ATP产生保持低水平。因此,摄入的卡路里被优先用于生成脂肪。随着时间的推移,ATP水平得到补充,但代价是脂肪储存的显著增加。
尽管大多数评估内源性果糖产生的研究都是在实验室动物上进行的,但有数据表明年轻瘦成人的内源性果糖产生可能超过每天5克,而在高血糖软饮料后,果糖产生率可能会增加两倍或更多。在高血糖、高盐或高糖饮食的受试者中,预计产量会更高,因为已知在这种情况下AR会被上调。高血糖和高盐饮食也可以在大脑中诱导果糖的大量产生。因此,果糖可以从饮食中获得和/或产生(糖、高果糖玉米糖浆、高血糖碳水化合物、咸食、鲜味食物、酒精),以及在压力条件下(缺血、缺氧和脱水)。实际上,三种吸引人的味道(甜、咸、鲜)都鼓励摄入产生果糖的食物,而苦和酸的味道可能是为了避免可能携带毒素的食物而发展出来的。
3. 果糖通过降低ATP水平触发生存开关
体重通常受到严格调节,尤其是对于野生动物而言。最强的调节器之一似乎是瘦肉(无脂肪)体质,这个测量值与能量摄入和静息能量代谢都有关联。动物试图保护的关键特征之一是肌肉质量并不奇怪,而这又由线粒体功能的水平指导。因此,动物试图保持细胞内ATP水平,因为这代表它们的活跃能量,并且它们通过具有代谢灵活性来实现这一点,其中任何消耗的ATP都可以从营养素摄入或脂肪储存中迅速替换(图1)。在这种情况下,以未被急性增加的能量消耗所补偿的水平施用高热量饮食将导致体重增加(和脂肪积累),而低热量饮食将导致体重减轻(和脂肪储存的耗尽)。在这两种情况下,能量平衡得以维持,ATP水平得以保留。
相比之下,果糖的代谢方式不同,尽管它也遵循能量平衡的规则。具体来说,果糖积极降低细胞内ATP水平的同时,还减少了生成新ATP的能力(图1)。因此,代谢灵活性被阻断。ATP水平下降到不至于威胁生存的程度,但足以激活一个警报,表明可用的能量储存有耗尽的风险。例如,在人类研究中,口服摄入果糖后,肝脏中的ATP水平可以下降20%,如果通过静脉给予,则可下降60-70%。ATP耗竭的水平与肝脏暴露于的果糖浓度有关,这与摄入量和吸收速度有关,后者在果糖以液体形式给予时更大。这触发了一系列生物学反应,导致能量摄入增加。然而,线粒体功能的持续抑制导致卡路里被转移到储存的能量(脂肪)中。最终ATP水平被替换,但代价是储存更多的脂肪,使得总体能量水平(即活跃和储存的能量)更高。
果糖降低细胞内ATP的具体机制如图2所示。果糖通过果糖激酶C(也称为酮己糖激酶-C,或KHK-C)迅速磷酸化为果糖-1-磷酸,该酶没有反馈系统来保护ATP水平,从而导致细胞内ATP和磷酸盐急剧下降。细胞内磷酸盐的下降刺激AMP脱氨酶-2(AMPD2),移除AMP底物,从而减缓ATP的再生,同时刺激尿酸的产生(从AMP降解为IMP以及从头嘌呤合成)。这有助于维持(AMP + ADP)/ATP的正常电荷比率。虽然一些IMP可能通过嘌呤挽救途径重新转换为AMP和ATP,但尿酸的产生似乎更受青睐,血清尿酸在摄入后的第一小时内可以增加0.3-2.0 mg/dl。
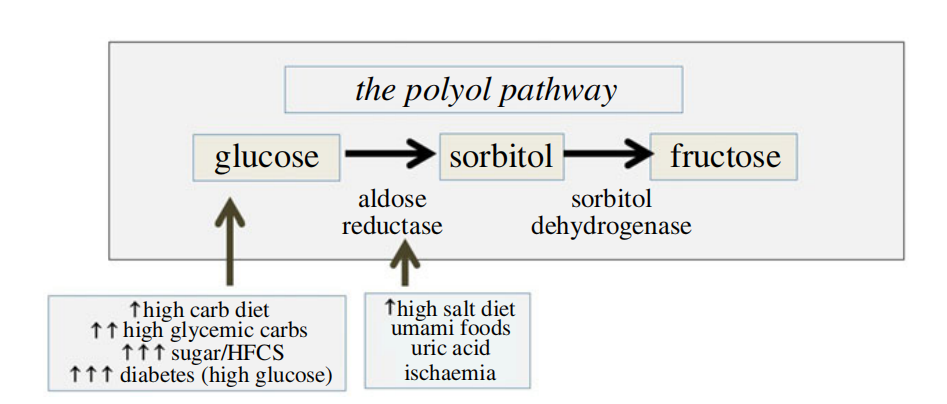
图2.内源性果糖的产生。果糖是通过多元醇途径从其底物葡萄糖产生的。醛糖还原酶是限速酶。果糖产生的主要驱动因素是高血糖水平,以及可以刺激醛糖还原酶活性的因素。从食物的角度来看,这包括高血糖碳水化合物、咸食、酒精和鲜味食物,如啤酒。然而,这条途径也可以通过压力激活,包括缺血、缺氧、脱水和热应激。
黄嘌呤氧化还原酶产生的尿酸会产生氧化剂(主要是过氧化氢),但尿酸本身也刺激NADPH氧化酶转移到线粒体。由于NADPH氧化酶和内源性线粒体氧化应激的增加,以及保护性抗氧化系统(特别是Nrf2)的同时减少,线粒体的氧化应激增加。氧化应激通过抑制柠檬酸循环中的顺乌头酸酶以及通过乙酰化肉碱棕榈酰转移酶-1a(CPT1α,参与脂肪酸运输到线粒体中)和烯酰CoA水合酶(β-脂肪酸氧化途径中的酶)阻断β-脂肪酸氧化,从而减少ATP产生,并通过AMP激活蛋白激酶阻断ATP再生。顺乌头酸酶的抑制刺激了参与脂质生成的酶,而微生物群对果糖的响应产生的乙酸酯用于生成乙酰CoA,提供了底物。此外,有一些证据表明果糖代谢可能导致烟酰胺腺嘌呤二核苷酸(NAD+)的消耗,降低NAD+/NADH比率并影响氧化还原平衡,导致sirtuins减少,这也可能赋予代谢效应并增强糖酵解反应。这可能是CPT1α乙酰化的原因。
虽然线粒体氧化磷酸化和ATP生成受到抑制,但果糖-1-磷酸的产生刺激了葡萄糖激酶从细胞核释放,导致葡萄糖摄取和糖原产生,而果糖-1-磷酸的分解产生了甘油醛和二羟丙酮磷酸盐。在禁食状态下,这刺激了糖异生,可以帮助提供葡萄糖作为能量底物,而在进食状态下优先刺激糖酵解。例如,基于放射性示踪剂研究,乳酸可能占摄入果糖的多达25%。虽然乳酸可用于生成乙酰CoA作为柠檬酸循环的底物,但当乳酸积累时,它会产生活性氧物种,损害脂肪酸进入线粒体并减少线粒体ATP生成。果糖对糖酵解的净刺激以及通过抑制氧化磷酸化减少氧气消耗很可能旨在为面临缺氧风险的动物提供生存益处。
这种降低细胞内ATP的伎俩似乎是激活生存反应和破坏体重调节的核心。实际上,为了纠正ATP的不足,卡路里的摄入受到刺激,但转换机制将卡路里转移到脂肪中。最终,ATP水平得到补充,但代价是增加脂肪堆积。随着时间的推移,由于线粒体的反复氧化应激导致永久性的线粒体功能障碍和损耗,情况会发生变化。现在,ATP水平一直保持低水平,但身体通过减少脂肪质量和降低静息能量代谢来适应低ATP水平。能量摄入必须保持低水平,否则体重会再次增加。
与假设降低细胞内ATP是肥胖和代谢综合征中一个重要的触发因素一致,果糖以及尿酸可以在多种细胞类型和组织中诱导ATP降低。果糖引起的肝脏ATP耗竭可以通过别嘌呤醇部分逆转。更重要的是,低细胞内ATP状态是肥胖、糖尿病、非酒精性脂肪肝病(NAFLD)和阿尔茨海默病的特征。
4. 果糖代谢诱导的“生存开关”的描述
给予果糖可以完全复制代谢综合征,并导致体重增加、内脏脂肪堆积、胰岛素抵抗、高三酸甘油酯血症、低高密度脂蛋白胆固醇、血压升高、脂肪肝、微量白蛋白尿、高尿酸血症和全身炎症的生物标志物(包括低脂联素、高瘦素和高敏感性C-反应蛋白)。这些发现也在准备冬眠的动物中观察到,表明“代谢综合征”这一术语可能用词不当,这些特征反而应该被描述为“脂肪储存综合征”。所有这些特征作为生存反应的一部分发展(图3),如下所述:
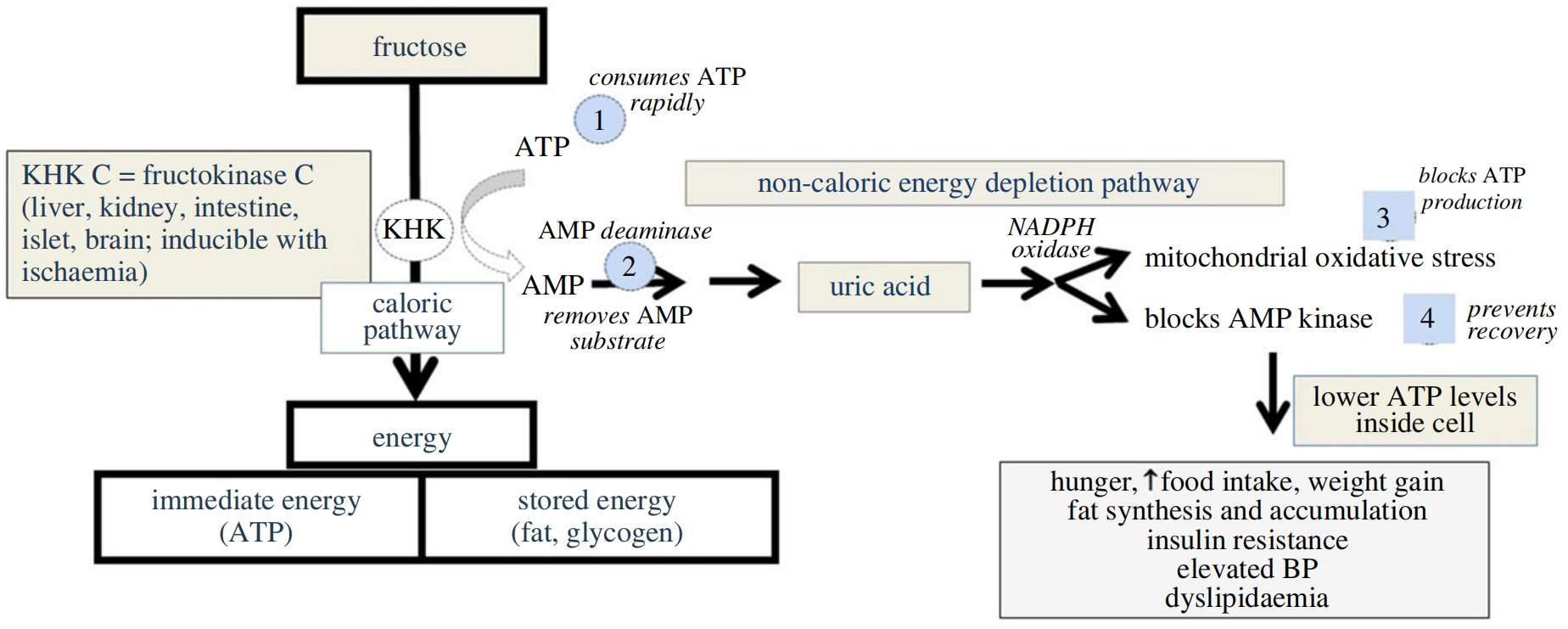
图3.果糖代谢及降低细胞内ATP水平的机制。果糖首先由KHK-C磷酸化为果糖-1-磷酸,导致ATP迅速消耗,这直接依赖于果糖的浓度。与葡萄糖不同,葡萄糖不会看到ATP显著下降,KHK-C驱动的反应可以使ATP和细胞内磷酸盐浓度骤降。反过来,低细胞内磷酸盐触发AMP脱氨酶-2(AMPD2)的激活。在ATP耗竭期间产生的AMP随后被代谢为肌苷一磷酸(IMP)并最终转化为尿酸。AMP的利用移除了一些再生ATP所需的AMP底物。然而,细胞内尿酸刺激转位到线粒体的NADPH氧化酶,同时抑制线粒体抗氧化防御机制,特别是Nrf-2。线粒体的氧化应激抑制了线粒体β-脂肪酸氧化(抑制烯酰CoA水合酶)和三羧酸循环(通过阻断顺乌头酸酶),导致氧化磷酸化产生的ATP受到抑制。此外,尿酸还抑制了在低能量状态下帮助再生ATP的AMP激活蛋白激酶(AMPK)。因此,果糖代谢KHK-C的净效应是降低细胞内ATP水平。
(a) 寻找食物和水
果糖鼓励行为改变以帮助寻找食物和水。这包括通过激活食欲素-下丘脑回路刺激饥饿和暴饮暴食行为,尽管这本身并不刺激增加食物摄入。果糖还可能通过将水分转移到与糖原生产相关的细胞内来增加血清渗透压,从而刺激口渴。最重要的是,果糖通过损害饱腹感来破坏正常的体重调节,导致过量的食物(能量)摄入。这需要几周的时间来发展,并由中枢(下丘脑)瘦素抵抗介导。果糖还作用于大脑以刺激觅食反应,包括刺激探索行为、冲动性和增加运动活动。通过提高尿酸也可以观察到类似效果。研究表明,这些效果是通过抑制大脑中涉及自我控制、近期记忆和深思熟虑的区域(如大脑皮层、内嗅皮层、海马和后扣带皮层)的胰岛素敏感区域介导的。
(b) 增加脂肪和糖原储存
除了通过诱导瘦素抵抗刺激过量卡路里摄入外,果糖还增加肠绒毛长度,可能有助于更有效的食物吸收。果糖代谢还刺激脂肪生成,损害β-脂肪酸氧化,并可能因发展高胰岛素血症而减少脂肪细胞的脂解。糖原储存也增加。
(c) 能量保存
尽管觅食反应需要消耗能量,但这通过降低静息能量代谢得到补偿,这可能与脂肪酸氧化的阻断和胰岛素抵抗减少肌肉中葡萄糖代谢有关。此外,骨骼肌和脂肪细胞的葡萄糖摄取减少有助于保持正常或高水平的血糖水平,从而保护那些对胰岛素依赖性较低的大脑区域。然而,大脑的某些区域更依赖于胰岛素,特别是那些涉及抑制觅食反应的区域。果糖损害了胰岛素信号传递到涉及自我控制、深思熟虑和近期记忆的区域。结果是在系统性和大脑胰岛素抵抗的帮助下刺激觅食同时帮助节省能量。
(d) 关键身体功能的保留
处于危险条件下的动物需要维持强健的循环和排泄功能。毫不奇怪,果糖代谢导致血压急性升高,这依赖于尿酸。肾脏功能通过增加肾小球滤过压来维持,实验性高尿酸血症也观察到类似的发现。果糖还刺激抗利尿激素的产生,有助于水的重吸收,并且果糖还促进肾脏近曲小管和肠道中的钠吸收。这些效应有助于生存(图4)。
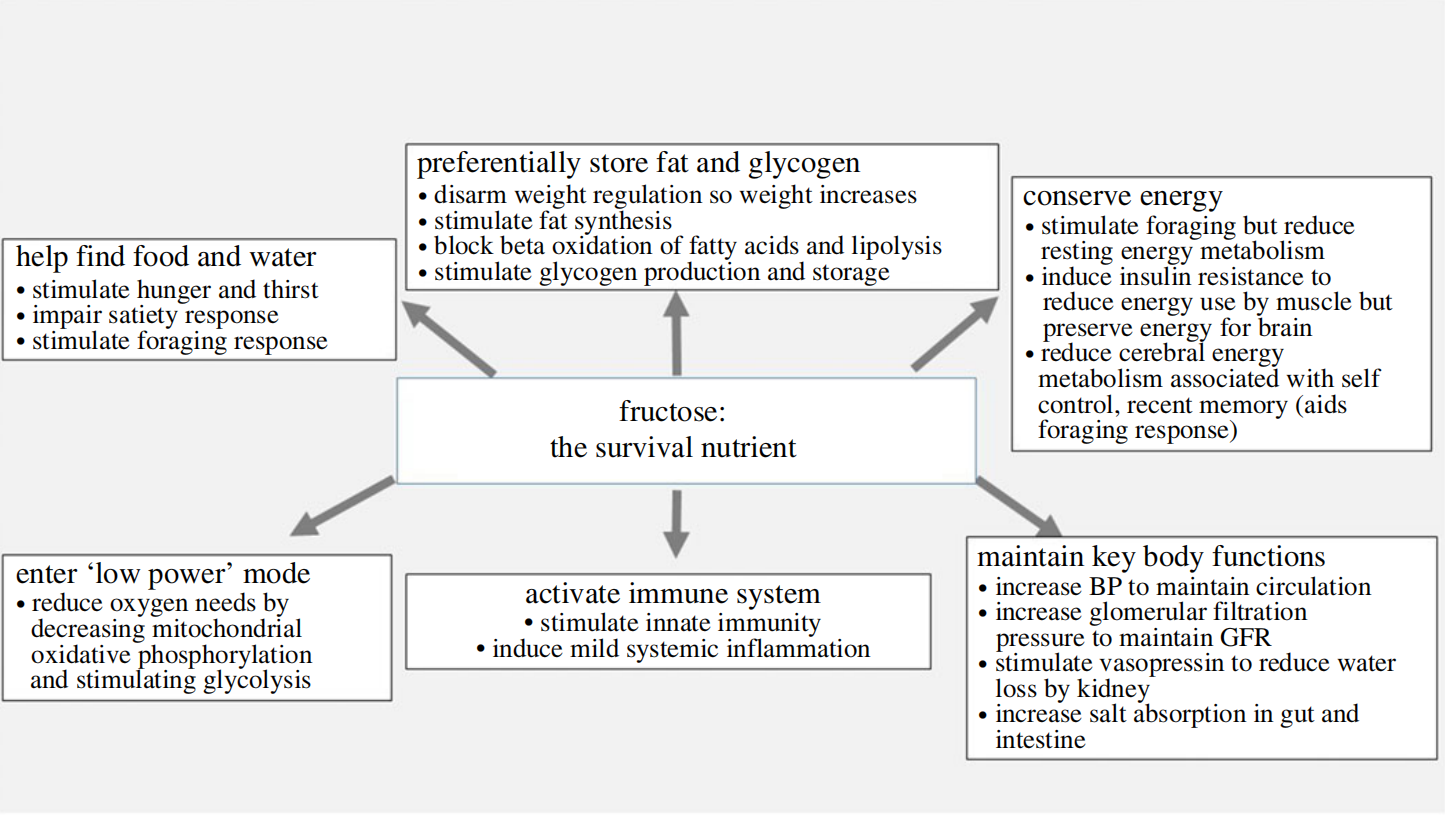
图4.对果糖的生存反应。
(e) 免疫系统的激活
果糖产生的尿酸与炎症途径的刺激相关,包括激活MAP激酶(p38)、NF-ΚB和刺激炎性体,以及协助树突状细胞功能。升高的尿酸刺激氧化应激以及趋化因子、C-反应蛋白和炎症途径的产生。再次,免疫激活提供了防止感染的保护。
(f) 进入低功率模式
果糖代谢引起的线粒体氧化磷酸化减少和糖酵解的刺激使身体进入低功率模式并降低其氧气需求。这在炎症区域局部缺血的情况下提供初始益处,而长期可能会驱动炎症和纤维化。例如,在心脏缺血时,会产生果糖,抑制线粒体氧化磷酸化并刺激糖酵解,从而驱动心脏重塑。这似乎部分依赖于转录因子HIF-1α的激活。线粒体功能降低与糖酵解的刺激也是糖尿病肾病的特征。我们已经报告说,糖尿病肾病是由肾脏中葡萄糖转化为果糖介导的。看来,SGLT2抑制剂在心脏和肾脏疾病中的益处可能是通过阻止葡萄糖进入病变组织,从而防止其随后转化为果糖。果糖代谢的阻断可能启动脂肪氧化和AMP激活蛋白激酶活性,类似于观察到的休眠动物。果糖降低线粒体氧化磷酸化并刺激糖酵解(Warburg效应)的效果解释了为什么癌细胞偏好果糖作为生长介质,以及为什么癌细胞生长受到果糖和尿酸的刺激。
5. 果糖代谢在肥胖和代谢综合征流行中的作用
肥胖和糖尿病的流行始于20世纪初。一个可能的机制与糖分摄入量的急剧上升有关,当引入高果糖玉米糖浆时出现急剧转折。含有果糖的软饮料和其他液体特别有效地激活该途径,因为ATP下降直接与肝细胞暴露的浓度有关,这不仅与果糖的数量有关,还与摄入速度有关。实际上,我们发现减慢含果糖苹果汁的摄入可以减少生存途径的激活。确实,含糖饮料的摄入与肥胖和代谢综合征的风险相关。同样,全球肥胖和糖尿病流行与糖分摄入量的增加以及将糖引入非西方文化有关。此外,加工食品中富含糖和盐的摄入量增加,以及高血糖碳水化合物和酒精的摄入也促进了果糖的摄入或内源性产生,以及肥胖、2型糖尿病和高血压的风险。
然而,有证据表明人类对果糖更敏感,因为老鼠和大鼠通常对果糖的影响相对抵抗,除非给予大剂量。在这方面,我们的小组已经确定了两个显著增加果糖诱导代谢综合征风险的基因突变。第一个是大约61百万年前发生的维生素C突变,这与结束白垩纪的小行星撞击事件时间相近。维生素C是一种抗氧化剂,可以阻断果糖效应,并已发现对代谢综合征的各个组分有有益效果。我们的小组发现,喂食维生素C缺乏的果糖小鼠表现出剂量反应,其中较高剂量的维生素C对于相同量的果糖摄入导致较少的肥胖(未发表的观察)。因此,维生素C突变可能在小行星碰撞后发生的“撞击冬季”期间为早期灵长类动物提供了自然选择优势。另一种可能使我们的灵长类祖先受益的突变是尿酸酶突变,这一突变从大约2400万年前开始逐步发生,直到中新世中期该基因完全消失。尿酸酶是一种降解尿酸的酶,缺乏尿酸酶的小鼠在摄入果糖后尿酸水平显著升高。我们已经讨论了证据,表明失去尿酸酶很可能代表了一个真正的“节俭基因”。这个突变发生在全球变冷导致祖先猿类几乎灭绝的时期。抑制尿酸酶可以显著增强果糖诱导大鼠代谢综合征的能力,这一事实证明它可能是一个节俭基因;而复活祖先的尿酸酶被证明可以阻止果糖在人类肝细胞中的生脂作用。值得注意的是,当突变发生时,它只是将血清尿酸水平翻倍(达到约3-4 mg/dl),因此主要起到防止饥饿的作用,而不是导致肥胖。然而,在20世纪,随着糖分摄入量的增加和肥胖、糖尿病及心血管疾病的上升,血清尿酸水平急剧上升。尿酸水平超过8 mg/dl的人更有可能发生肥胖和代谢综合征,实验和初步研究表明尿酸在其中起着促进作用。
6. 果糖如何导致体重增加?
观察到果糖刺激食物摄入以及降低静息能量代谢表明,能量摄入增加和能量消耗减少都可能是导致肥胖的原因,这通过体重和脂肪质量的增加来确定。然而,静息能量消耗的减少部分被觅食反应中增加的能量使用所补偿。
为了评估这两种机制的重要性,我们进行了配对喂养研究,其中给予大鼠高果糖食物(在某些情况下,还有糖),并与成分和热量含量相同但用淀粉替代果糖的饮食进行比较。在一些研究中,动物被置于热量限制下,但每组摄入的热量相等。主要发现是,摄入相同热量的动物无论饮食中是否含有果糖,体重变化相似。尽管摄入糖或果糖的动物体重略有增加(这可能是由于静息能量代谢降低),但总体体重变化不显著。因此,至少在为期四个月或更短时间的研究中,体重增加主要由热量摄入增加引起。
为了确定是什么驱动了能量摄入增加,我们进行了额外的研究。首先,我们发现甜味鼓励果糖摄入,但缺乏味觉的小鼠仍然偏爱果糖与水相比,并且随着时间的推移变得比对照组更胖。对果糖的偏好依赖于果糖的代谢,因为缺乏果糖激酶的小鼠对果糖的喜爱很少,尽管它们仍然喜欢蔗糖,可能是因为其葡萄糖含量。重要的是,基于我们在特定器官中敲除果糖激酶的研究,对果糖的偏好并不是导致热量摄入增加的原因。实际上,对果糖的偏好是由肠道果糖激酶介导的,而代谢综合征是由肝脏果糖代谢驱动的。重要的是,肝脏特异性KHK敲除小鼠会大量饮用果糖,但完全免受体重增加和代谢综合征的影响。
能量摄入增加的主要机制是瘦素抵抗的发展。最初,喂食蔗糖或果糖的动物减少了它们的常规食物摄入以维持中性能量平衡,因此不会发生体重增加。然而,几周后,动物增加了它们的常规食物摄入,使得总热量摄入变得高热量,这与体重增加相关。研究表明,这是由于中枢瘦素抵抗的发展,并且可以通过阻断果糖代谢来预防。事实上,我们发现这一机制特别由果糖激酶C亚型介导,并且是通过侧链反应驱动能量耗竭。
准备冬眠的动物表现出生存开关的类似特征(例如,过度进食、瘦素抵抗、体重增加和脂肪积累、脂肪肝和胰岛素抵抗),但在它们冬眠前不久,即使食物仍然可用,它们也会减少食物摄入,这与它们的代谢降低有关。一个可能的机制可能与达到触发减少食物摄入的生物学反应的重量有关(重力计)。虽然这种机制仍然发生在肥胖的人身上,但显然可以被持续暴露于高浓度果糖所超越,我们推测这可能与继续接触高浓度果糖有关。
(a) 脂肪的作用
瘦素抵抗似乎由摄入或内源性果糖驱动,而其他食物如高脂肪饮食则不会引起瘦素抵抗。虽然果糖介导瘦素抵抗,但在已经变得瘦素抵抗的动物中,高能量密度、高脂肪食物的摄入放大了体重增加。
确实,阻断瘦素信号会刺激对高脂肪食物的偏好,这是有道理的,因为它有利于更快地增加体重和脂肪积累。地松鼠在积极增重准备冬眠前,会优先食用含有高多不饱和脂肪的种子。此外,最近的研究显示,对脂肪的喜好是由TRPM5味觉受体以及肠道-迷走神经通路介导的,这些通路驱动了对脂肪的偏好。这就是为什么脂肪与果糖或糖的结合比单独的脂肪或果糖更能显著增加脂肪积累和体重增加。此外,西方饮食中的人们可能摄入了足够的糖、盐、高血糖碳水化合物和酒精,使大多数人产生了一些瘦素抵抗。例如,我们报告说,即使在低糖含量的情况下,内源性果糖也能促使喂食类似西方饮食(50%碳水化合物)的小鼠出现老化变化——缺乏果糖激酶的小鼠保持苗条和健康。这些发现也解释了为什么因纽特人尽管饮食高脂肪、高蛋白质却保持苗条,以及为什么“低碳”饮食尽管高脂肪却不会导致体重增加。例如,猪油不会使正常小鼠体重增加,除非它们首先被果糖诱导产生瘦素抵抗。
(b) 葡萄糖的作用
我们摄入的大部分果糖来自添加的糖(蔗糖和高果糖玉米糖浆),这些也含有葡萄糖。葡萄糖可以显著增强果糖的吸收,从而加剧果糖效应。然而,也有证据表明单独给予的葡萄糖也可以诱导肥胖和代谢综合征。一个流行的假设是,添加的糖和高血糖碳水化合物可能通过过度刺激胰岛素而导致肥胖,这不仅刺激脂肪的储存,还阻止脂肪细胞中的脂解。然而,如前所述,高血糖食物还可以激活多元醇途径并产生内源性果糖。
为了评估葡萄糖-胰岛素轴在驱动肥胖中的作用,我们给KHK-KO小鼠或对照小鼠施用了葡萄糖。对照小鼠表现出显著的体重增加、脂肪肝和胰岛素抵抗,但也在其肝脏中有高水平的果糖。缺乏KHK的小鼠喝了相同量的葡萄糖但吃得更少,虽然它们发展出了轻度肥胖,但大约只有野生型对照组的一半。KHK-KO小鼠还有最小的脂肪肝或胰岛素抵抗。这些研究因此证明,葡萄糖可能通过反复刺激胰岛素导致一些肥胖,但高血糖碳水化合物导致肥胖和代谢综合征的一个主要机制是通过内源性果糖的产生。后来当我们给缺乏KHK的小鼠施用高果糖玉米糖浆时,进一步的支持出现了,我们发现它甚至更能防止肥胖、脂肪肝和胰岛素抵抗,其效果大约是葡萄糖-胰岛素通路的四分之一。因此,尽管碳水化合物-胰岛素模型仍然是驱动肥胖的一个重要机制,果糖的转化在高血糖碳水化合物如何导致肥胖中扮演了重要角色。有趣的是,果糖诱导的胰岛素抵抗导致空腹胰岛素水平升高,这起到抑制脂解的作用,因为脂肪细胞在胰岛素抵抗状态下仍对胰岛素的抗脂解作用敏感。因此,果糖诱导的胰岛素抵抗状态也支持了葡萄糖-胰岛素假说,但内源性果糖是一个中间步骤。
(c) 蛋白质的作用
蛋白质对于维持精瘦体质量至关重要,有一些证据表明低蛋白饮食可能会刺激增加能量摄入以达到这一目标,但结果是增加了体内脂肪储存(蛋白质杠杆假说)。然而,大多数低蛋白饮食都是高碳水化合物饮食,这使得很难知道在驱动肥胖中哪个更重要,低蛋白饮食还会刺激FGF21,通过刺激脂肪氧化来抵消果糖的影响,尤其是在肝脏中。
此外,某些蛋白质,如红肉和加工肉类,可能会增加患糖尿病、肥胖、痛风和慢性肾病的风险。一个可能的解释是,红肉和贝类富含谷氨酸和核苷酸,如IMP和AMP。IMP是果糖能量耗尽途径的关键成分,谷氨酸也在肝脏中代谢为尿酸。实际上,我们发现鲜味(谷氨酸和IMP)可以产生尿酸并导致肝ATP耗尽、瘦素抵抗以及类似果糖的肥胖和代谢综合征。用别嘌醇阻断尿酸的产生可以阻止单钠谷氨酸引起肥胖的效果。谷氨酸导致尿酸产生的机制可能与其转化为谷氨酰胺和新尿酸的从头合成有关。已知痛风患者往往血清谷氨酸水平较高,并且也会因富含谷氨酸的食物(如番茄)而发展成痛风。然而,大多数富含嘌呤的蔬菜不太可能提高尿酸并引起痛风,可能是因为它们的嘌呤相对缺乏AMP和IMP。
有趣的是,提高尿酸的富含嘌呤和谷氨酸的食物往往是鲜味食物,这表明这些是动物倾向于寻找的食物。尿酸通过刺激线粒体氧化应激和抑制AMPK的能力直接激活开关,但它也可以刺激果糖产生并增强KHK表达。因此,许多效果是通过刺激从葡萄糖产生果糖来解释的,这可能解释了为什么高蛋白质饮食在没有碳水化合物的情况下不会导致肥胖。
7. 果糖和代谢综合征:一种与过量热量无关的效应
我们配对喂养研究的一个显著发现是,尽管体重增加是由过量的热量驱动的,但果糖的其他效应即使在热量摄入受限时也会发生。事实上,在一项研究中,给大鼠喂食低热量、高蔗糖的饮食,这些蔗糖喂养的大鼠仍然发展出了严重的脂肪肝、高三酸甘油酯血症、胰岛素抵抗和血压升高。我们还发现,这些大鼠最初发展出高胰岛素血症,血糖水平正常,但随着时间的推移,它们表现出胰岛素逐渐下降,并伴随着明显糖尿病的发展。胰腺岛细胞也显示出玻璃样变和炎症,以及岛细胞上尿酸转运蛋白的从头表达。喂食淀粉的控制动物也出现了一些轻度高三酸甘油酯血症和胰岛素抵抗的迹象,我们现在认为这是由于低级别的果糖生成所致。然而,两组之间的整体差异是显著的。这些研究证实,代谢综合征,包括导致脂肪增加的身体成分改变,可以与最小限度的体重增加相关联。事实上,非酒精性脂肪肝病(NAFLD)可以发生在瘦人中,特别是如果存在高尿酸血症。
8. 盐、脱水和抗利尿激素在肥胖中的意外作用
水是一种关键资源,果糖增加血液中抗利尿激素水平作为增加水分保留的潜在机制并不令人惊讶,这是通过依赖抗利尿激素的尿液浓缩来实现的。我们还发现,果糖可能在调节抗利尿激素方面具有基本作用,因为脱水(高渗透压)被发现能激活视上核中的多元醇途径,并与抗利尿激素合成增加有关,而KHK-KO小鼠对急性和慢性脱水的抗利尿激素反应减弱。
一个重大惊喜是抗利尿激素不仅仅刺激肾脏中的水分重吸收,而是在果糖下游驱动生存开关的大多数特征。多年来,人们已经知道有另一种称为V1b受体的抗利尿激素受体,但其生理功能并不十分清楚。我们发现,V1b受体敲除小鼠完全免受果糖诱导的代谢综合征的影响。虽然确切的机制尚未完全理解,但我们确实知道V1b受体刺激促肾上腺皮质激素(ACTH)和胰高血糖素水平,并且可能还调节KHK的表达。
回顾过去,已知脂肪被海洋和沙漠哺乳动物用作代谢水的生成来源。动物在冬眠、夏眠时依赖于代谢水,长途迁徙的鸟类也是如此。摄入盐可以引发高渗透压和类似脱水的状态,而不损失水分,这也可以激活果糖和抗利尿激素的产生,并在实验室小鼠中引起依赖于果糖激酶的肥胖和代谢综合征。盐的摄入量在肥胖受试者中较高,并预测代谢综合征、糖尿病和脂肪肝的发展。大多数肥胖者也显示出脱水迹象并有高抗利尿激素水平(通过测量血清中抗利尿肽来注意)。用水补水以抑制抗利尿激素水平也可以部分停止甚至逆转代谢综合征的特征。这些研究表明,生存开关与营养素(果糖)、代谢废物(尿酸)和激素(抗利尿激素)紧密相连,肥胖确实是一种激素失调。
9. 生存开关的短期和长期健康后果
持续激活果糖介导的开关的最严重后果不是肥胖和体重增加,而是涉及西方社会许多常见疾病的代谢效应(表1)。这不仅包括与肥胖相关的经典疾病,如糖尿病、非酒精性脂肪肝病和痛风,还包括高血压、冠状动脉疾病、某些癌症、行为障碍和痴呆。
表1.生存开关的过度激活可能会驱动代谢疾病。
一个例子是该途径在推动冠状动脉疾病中的作用日益显现。最近,人们认识到全身性炎症以及动脉粥样硬化斑块中的炎症在冠状动脉疾病和心肌梗塞中发挥作用。一个潜在的机制是可溶性尿酸激活炎症体和NFkB介导的炎症作用。然而,最近发现了一个更戏剧性的机制,因为85%的痛风患者在其血管中有尿酸晶体,这些晶体似乎集中在动脉粥样硬化斑块中。事实上,高尿酸血症和痛风与心血管死亡率的增加有关,这一点通过流行病学研究和孟德尔随机化研究都得到了证实。
另一个例子是衰老。果糖代谢与氧化应激、线粒体功能障碍、细胞保护转录因子核因子红细胞2相关因子2(Nrf2)的丧失以及表征衰老过程的sirtuins减少有关。果糖还比葡萄糖更有效地诱导高级糖基化终产物的生成。如前所述,我们发现与衰老相关的肾脏疾病在衰老的KHK-KO小鼠中没有发生。
还有越来越多的证据表明,阿尔茨海默病可能与大脑内部的果糖代谢有关。饮食中富含含或产生果糖的食物(高糖、高盐、高血糖和高加工红肉饮食)的人,或者具有代谢综合征和糖尿病特征的人,患阿尔茨海默病的风险增加。给予果糖也可以在实验室大鼠中诱导出阿尔茨海默病的特征(包括精神状况下降和淀粉样蛋白及τ蛋白沉积),并且在早期阿尔茨海默病痴呆的大脑中果糖也升高(综述于)。机制似乎是由于过量果糖的生物学效应,抑制大脑的某些区域以刺激觅食反应。
一个关键发现是,果糖途径的作用几乎不可避免地在早期疾病状态中最强烈,因为随着时间的推移,常常会出现纤维化、炎症或线粒体丢失,导致疾病过程持续存在(表1)。因此,干预的最佳时机可能最终是在早期疾病阶段,此时条件还未变得不可逆转。尽管如此,由于许多疾病代表了线粒体功能受抑制的代谢障碍,一种理想的方法是尝试通过运动或其他方式再生线粒体。在这方面,虽然低果糖和低盐饮食都能改善线粒体功能,但另一种极好的方法是通过运动。
10. 限制和挑战
这一假设面临挑战。首先,关于节俭基因假设是否存在一直有争议,因为大多数饥荒时间过短,无法提供足够的自然选择压力,导致决定物种命运的生存基因出现(讨论于)。一些人还争论说,如果节俭基因存在,那么今天每个人都应该肥胖。然而,尿酸酶的突变发生在持续数百万年的季节性饥荒期间,而且这种突变并没有导致肥胖,而是防止了饥饿。我们的假设也表明,并不是每个人都会发生肥胖,因为这需要一种相互作用,即一个人因果糖代谢而变得瘦素抵抗,然后摄入高能量食物(如脂肪)。
我们同意,在现代人类时期,生存突变的出现可能性要小得多。话虽如此,在冰河时期的5000到10000年间,前进的冰川和大型猎物可用性的减少导致了间歇性的饥饿和人口收缩,这影响了萨满教和艺术的发展。在4.2千年前左右还有一场持续200年的干旱,导致多个王国的崩溃,并导致了乳糖持久性基因的迅速传播,因为它具有生存优势。然而,现在生存基因出现的可能性要小得多,增加的脂肪储存不太可能带来好处,因此在今天的文明社会中,肥胖基因可能更多地通过随机漂移传播(漂移基因假说),特别是因为对于肥胖和久坐的人来说,捕食风险较小。
还有一些证据表明,过去十年糖分饮料的摄入量有所下降,导致添加糖的总体摄入量减少。然而,添加糖的摄入量仍然很高,仍占总体饮食摄入量的15%。加工食品中有超过70%的产品含有过多的添加糖和盐。因此,很可能大多数人接触到的含有或刺激果糖产生的食品仍然超过了诱导瘦素抵抗所需的阈值。
还有一些证据表明,脂肪本身可能在没有果糖的情况下驱动肥胖。我们注意到,饱和脂肪(黄油)可以在缺乏果糖激酶的小鼠中引起一些适度的肥胖,这与轻度肝脏脂肪变性有关。与其他脂肪相比,饱和脂肪似乎能更好地诱导肝脂肪变性,因为它们能够抑制脂解并增加游离脂肪酸向肝脏的输送。另一项研究发现,高动物脂肪饮食(特别是黄油和奶酪)可能会增加糖尿病的风险。尽管如此,因纽特人以肉类和饱和脂肪为主的饮食中缺乏肥胖和糖尿病,让我们认为这不是驱动肥胖和胰岛素抵抗的主要机制。事实上,上个世纪饱和脂肪的摄入量下降了,并且与肥胖、糖尿病和非酒精性脂肪肝病的流行不相关,而非酒精性脂肪肝病与含糖饮料的摄入有关。
另一个令人困惑的发现是,低碳水化合物、生酮饮食的受试者经常因为酮尿症干扰尿酸排泄而发展为高尿酸血症。虽然高尿酸偶尔会引发痛风,但尚不清楚高尿酸血症是否有其他有害影响。有人认为它可能有助于刺激氨基酸的糖异生,但也可能是因为酮血症的对抗效应或因为在低碳水化合物饮食的情况下由尿酸诱导的果糖生成不太可能。我们确实知道,在饥饿情况下,以及很可能在严重的碳水化合物限制下,果糖不再刺激脂肪储存,而是优先转化为葡萄糖以满足即时能量需求。显然,需要进行更多研究来解决生酮饮食中高尿酸血症的作用。
最后,众所周知,并非所有冬眠动物都摄入果糖作为触发事件来增加它们的食物摄入,一些在冬眠穴中的动物可能吃相同的食物却仍然经历一个明显的转换,其中它们大幅增加脂肪储存。这些动物中的触发因素尚不清楚,尽管它可能与水分状态的改变、食物不安全、遗传因素或其他未知机制有关。我们确实知道,尽管进食相同的饮食,进入冬眠的地松鼠在肝脏中显示出与果糖代谢相似的代谢变化,其中体重增加与肝脏中AMP脱氨酶和尿酸的激活有关,随后在冬眠期间氧化脂肪时激活AMP活化蛋白激酶。显然,需要更多的研究。
11. 可测试的预测
尽管果糖生存假说有很多支持性证据,但也有可测试的预测。首先,迫切需要确认内源性果糖途径在人类群体中的重要性,并确定其对生物学结果的影响程度。一种可能的方法是开发可以干扰该途径的药物,如开发有效的KHK、AMP脱氨酶-2或其他生存途径中酶的抑制剂。研究冬眠哺乳动物和长距离迁徙鸟类中内源性果糖途径的作用也将有所帮助。第二步是更好地理解从早期更可逆状态到更永久状态的转变机制。一种方法是识别记录这种转变的生物标志物。例如,通过评估风险综合征患者的初始检查中的血清抗利尿肽作为抗利尿肽途径重要性的最有效测试,并进行一项研究,在该研究中通过水合和盐分及糖分限制或使用抗利尿肽1b受体阻滞剂来抑制它。同样,研究尿酸作用的研究可能最有可能在疾病的早期阶段显示对代谢特征的益处。
12. 总结
肥胖及其相关的代谢疾病对现代社会造成了毁灭性的健康后果。在这里,我们将这些疾病与自然界中发展起来的一个主要生存途径联系起来,以帮助动物为稀缺时期做准备。不幸的是,我们不知不觉中在我们的日常饮食中采纳了激活这个开关的食物,加上我们获得的节俭基因,我们现在正在承受着将这个生存途径过度驱动的后果。我们的成功悲剧甚至比想象的还要大,因为新的研究表明果糖途径还可能增加我们患癌症、妊娠相关疾病和神经系统疾病的风险。我们建议进行上述概述的研究,以更好地了解果糖代谢在健康和疾病中的作用。
https://blog.sciencenet.cn/blog-41174-1440315.html
上一篇:衰老神经元驱动的大脑衰老
下一篇:氢气+汉防己碱治疗矽肺动物实验
