博文
AI 筛选 + 虚拟筛选:新药研发的 “双引擎” 的核心逻辑_MCE(MedChemExpress)
|
SBVS & LBVS
虚拟筛选也分“流派”: 一边是盯着蛋白结构精准“贴贴”的 SBVS,一边是靠小分子“撞脸认亲戚" 的 LBVS。但是!!!俩“门派”的干活思路完全不一样: 一个靠“精准匹配”,一个靠“找相似款”......
实际筛药时到底该 pick 哪个? 来,用大白话给你扒得明白!
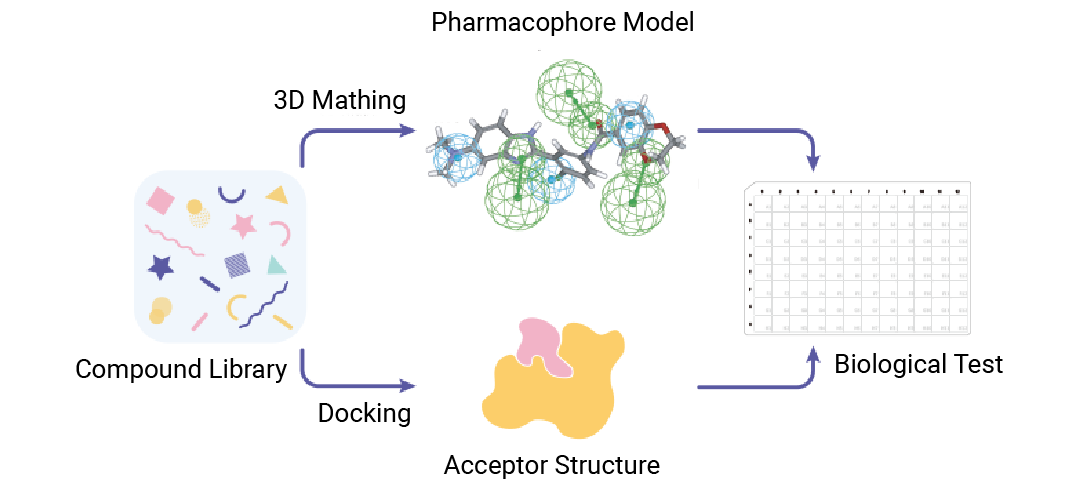
Section.01
什么是 SBVS ?
基于蛋白结构的虚拟筛选 (Structure-Based Virtual Screening,SBVS) 是借助靶标蛋白的三维结构,从化合物库中快速筛选出可能与蛋白结合的候选分子的计算药物设计方法,核心是 “利用蛋白的空间结构特征匹配潜在配体”。
因此,蛋白需要有确定可靠的结构是进行基于结构的虚拟筛选的前提。
核心逻辑:“锁钥模型”精准匹配
基于蛋白的活性口袋 (锁芯) 有特定的空间形状、电荷分布等特征,SBVS 通过计算化合物 (钥匙) 与蛋白活性口袋的 “匹配度” (比如分子对接) ,筛选出能稳定结合的化合物。
案例速览:基于 ZBP1 蛋白结构的虚拟筛选
面对“新颖、已知活性分子较少”的 ZBP1 蛋白 (与炎症、凋亡相关) ,复旦大学中山医院团队用 SBVS 实现突破:
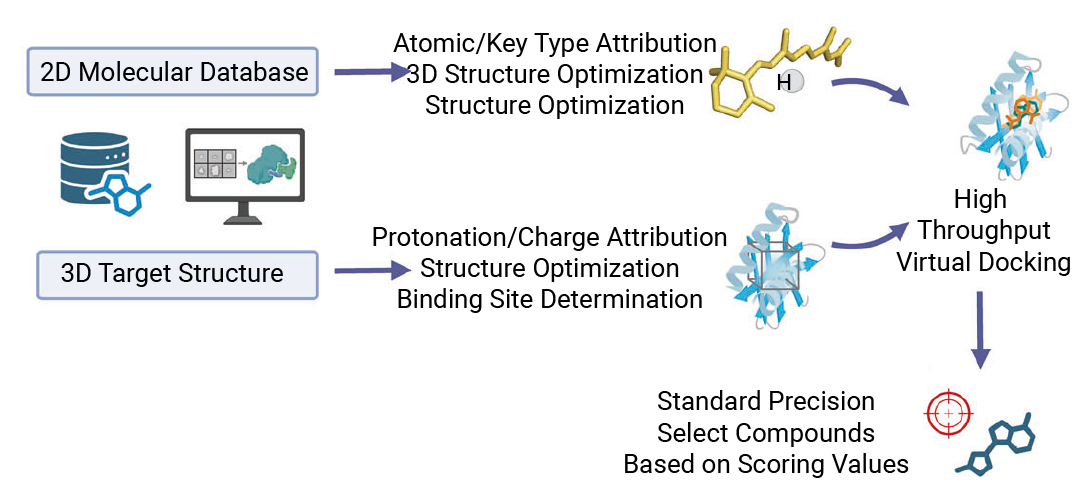
图 1. 小鼠 ZBP1 抑制剂筛选策略示意图[1]。
『1. Alphafold3 精准构建靶点模型』
ZBP1 的 RHIM 结构域是其与 RIPK3、CASP8 等蛋白相互作用的关键,但缺乏高分辨率晶体结构。研究人员用 AlphaFold 构建小鼠 ZBP1 三维模型 (置信度较高、可靠性良好) ,再借助 Schrödinger 软件分析,预测出 5 个潜在结合区域。其中,覆盖 RHIM 结构域大量关键氨基酸残基的“口袋 4”,被确定为核心筛选靶点——化合物结合此处,有望阻断 ZBP1 与其他蛋白的相互作用,抑制泛凋亡小体形成。
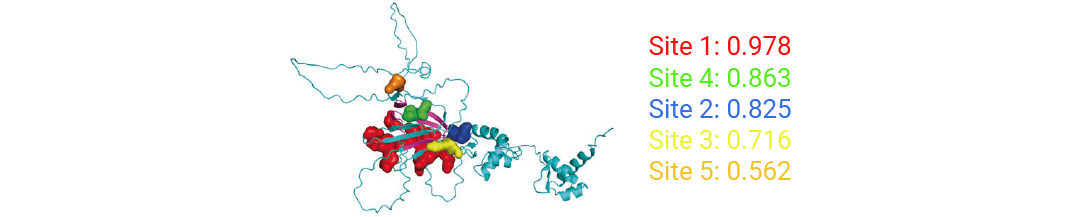
图 2. 基于小鼠 ZBP1 的三维结构,预测并评估其可成药结合口袋。根据位点 4 进行虚拟筛选[1]。
『2. 30 万个化合物高效对接』
随后,研究人员从 MedChemExpress (MCE) 化合物库选取 30 万个小分子,通过“高通量虚拟对接”模拟与 ZBP1 口袋 4 的结合过程。结合能越低,化合物与靶点结合越稳定、活性潜力越强。团队按结合能评分排序,最终筛选出 10 个结合能力最强的候选化合物。
『3. 验证活性:实验验证锁定 MSB』
虚拟结果需实验验证才可靠。研究人员用表面等离子体共振 (SPR) 技术检测:50μM 浓度下,MSB 与 ZBP1 的结合响应值 (RU=42.2) 显著高于其他化合物;进一步浓度依赖性实验显示,二者解离常数 (KD=725nM) 达“中高亲和力”水平,证实稳定结合。后续机制实验更验证,MSB 能有效阻断 ZBP1 与 RIPK3、CASP8、CASP6 的相互作用,完美实现筛选目标。
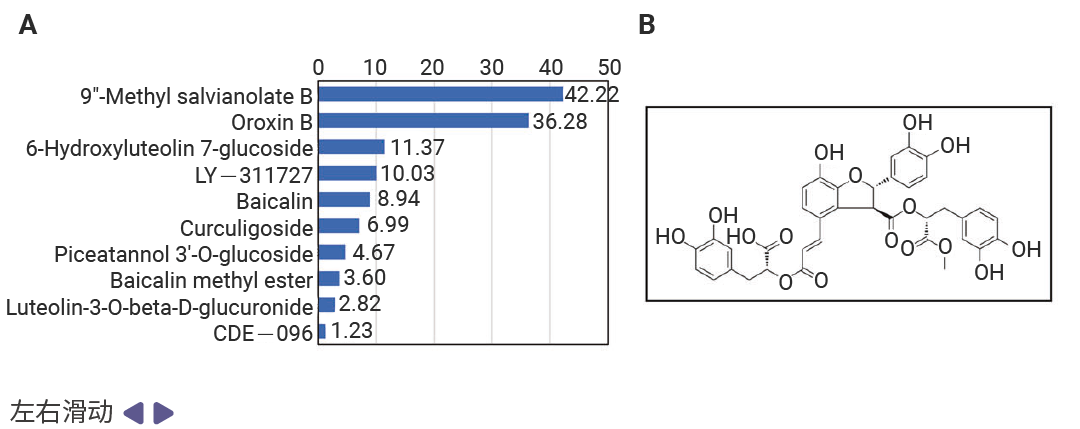
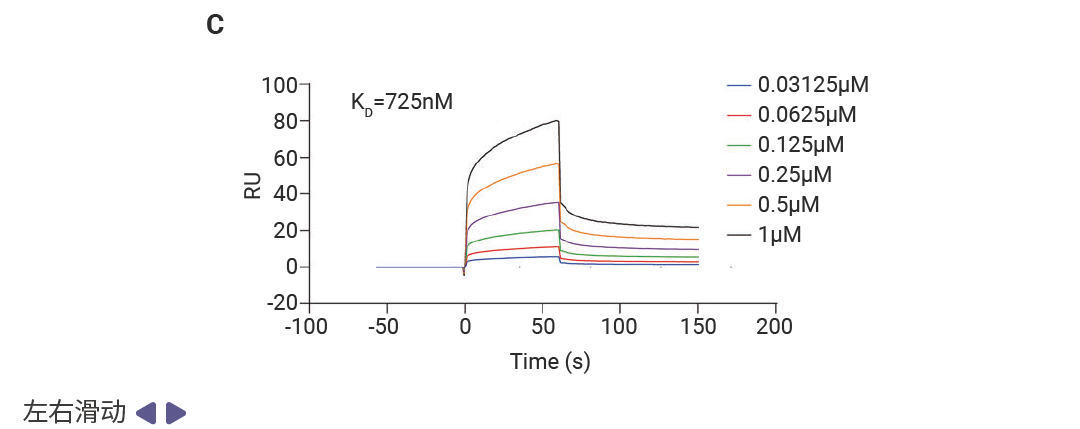
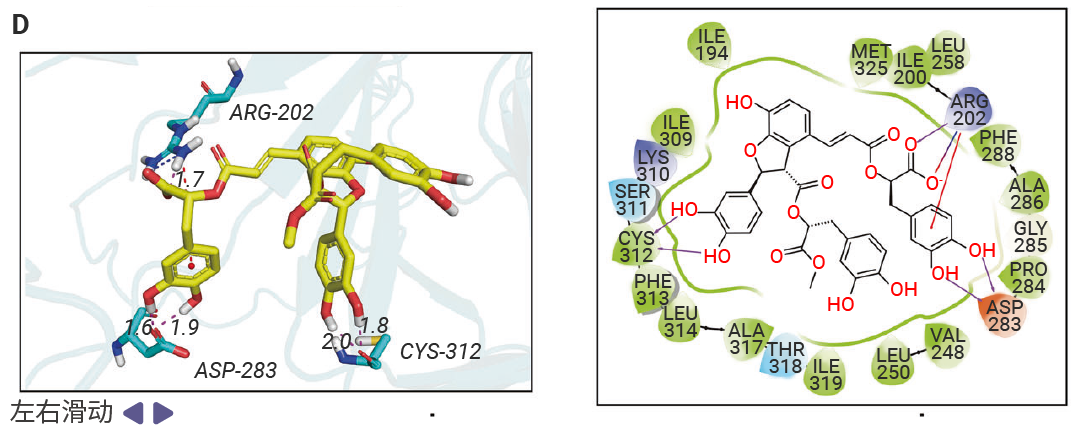
图 3. MSB 是 ZBP1 蛋白的先导结合分子[1]。
A.在单一浓度 (50 µM) 下,对十种化合物与 ZBP1 的结合反应进行表面等离子共振 (SPR) 分析。B. MSB的化学结构。C. 后续浓度依赖性 SPR 分析得到的动力学曲线,显示 MSB 与 ZBP1 的结合。D. MSB与小鼠 ZBP1 结合的二维和三维模式。
Section.02
基于配体的
虚拟筛选 (LBVS)
基于配体的虚拟筛选 (Ligand-Based Virtual Screening,LBVS) 是借助已知活性配体的结构/性质特征,从化合物库中筛选出具有相似特征的潜在活性分子的方法——核心是 “通过已知配体的‘共性’找新配体”。
核心假设:“结构相似,活性相似”
已知能和靶标结合的配体 (活性分子) 通常有相似的结构、药效团 (决定活性的关键基团) 或理化性质,LBVS 通过比对这些特征,从化合物库中找出 “和已知活性配体像” 的分子。
主要方法有相似性搜索、定量构效关系、药效团筛选等。
相似性搜索:将分子转化为二进制指纹 (如 ECFP4) ,用 Tanimoto 系数计算相似度,快速初筛超大型数据库;
定量构效关系 (QSAR):用多个已知活性化合物训练模型,预测新化合物的活性;
药效团筛选:提取已知活性分子的关键特征 (如氢键供体/受体、疏水基团) ,构建“药效团模型”,筛选符合特征的分子。
LBVS 通常有以下核心流程:(1) 收集已知活性配体:获取能与靶标结合的化合物 (比如已报道的抑制剂) ;(2) 提取特征:为参考配体和数据库化合物计算描述符 (1D:分子量、LogP;2D:指纹;3D:形状、场效应等) ;(3) 评估相似度/预测活性:用形状比对、机器学习等算法计算得分;(4) 排序输出:按相似度或预测活性排序,筛选候选分子。
案例速览:基于 CYP3A4 配体的 AI 虚拟筛选
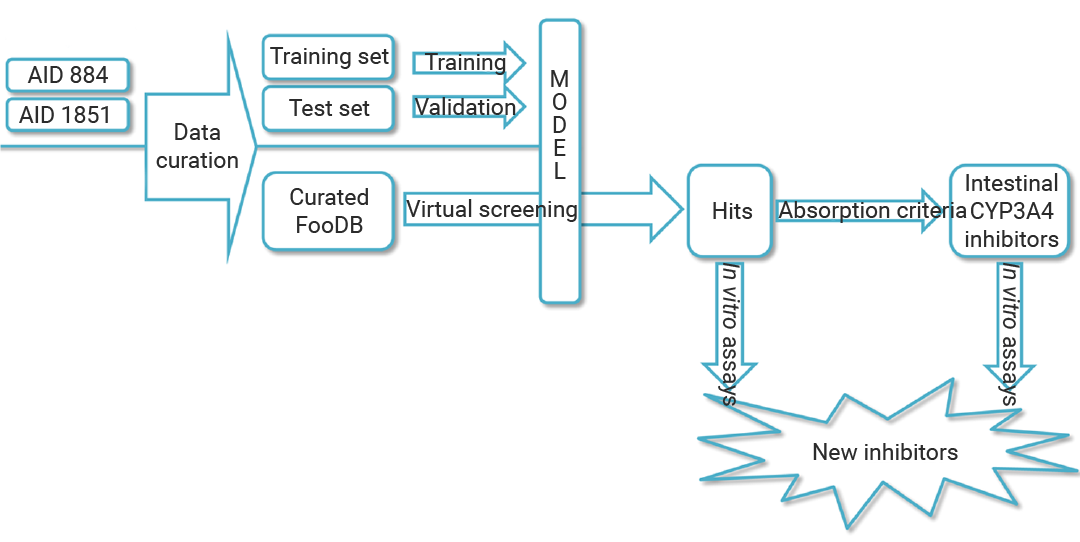
图 4. 基于配体的 CYP3A4 抑制剂筛选[2]。
耶路撒冷希伯来大学团队针对 CYP3A4 膳食抑制剂,构建“数据驱动建模-虚拟筛选-体外验证”三阶策略,效率与准确性双在线。
『1. 高质量数据集建模,训练“识别系统”』
首先,研究人员从 PubChem 获取 AID884、AID1851 高通量筛选数据,以 CYP3A4 介导的荧光素衍生物脱烷基化发光信号为活性指标,严格清洗数据 (移除片段化化合物、处理重复活性) ,最终得到 1760 个活性化合物 (IC500.032-15.85μM) 和 8893 个非活性化合物。基于 DeepChem 模块构建分类器,按 75%:25% 划分训练/验证集,以 AUC (0.97) 、特异性 (0.997) 、MCC (0.7) 优化,设定预测指数≥0.7 为活性阈值,同时用 Tanimoto 距离定义适用性域 (APD) ,排除不可靠预测。
『2. FooDB 化合物库筛选,精准过滤』
随后选取 FooDB 数据库 70474 个膳食化合物,先排除无机物、农药及药物残留,保留 68900 个独特分子;用建好的深度学习模型 (chemprop) 筛选,得到 136 个初始活性化合物,经 APD 验证和人工审核,最终锁定 115 个候选抑制剂。为聚焦肠道局部抑制剂,创新性结合 Lipinski 五规则,筛选出违反 2 项及以上规则 (如 MW>500、AlogP>5) 的 17 个低吸收化合物——这类分子更易在肠道发挥局部作用,减少全身干扰。
『3. 体外验证,证实模型可靠性』
最后,团队选取预测指数较高的银杏双黄酮 (0.81) 与苦鬼臼毒素 (0.83) ,构建大肠杆菌双顺反子表达系统,共表达 CYP3A4 与 POR 蛋白并提取微粒体;用荧光底物 Vivid BOMR 检测不同浓度化合物对 CYP3A4 活性的抑制率,通过非线性回归计算 IC50(分别为 12.9μM、3.5μM) ,成功验证二者活性,证明模型预测有效。
Section.03
SBVS + LBVS 联用
SBVS 和 LBVS 从不是“二选一”的对手,而是互补的“黄金搭档”: SBVS 依托靶点三维结构,通过分子对接精准预测小分子与活性口袋的结合模式,特异性强却受限于靶点结构解析难度;LBVS 无需靶点结构,凭借已知活性分子的相似性特征快速初筛化合物,适用性广却易局限于传统分子骨架。
二者联用可实现“初筛 + 精筛”的高效配合:先用 LBVS 缩小化合物库范围,再以 SBVS 提升筛选精准度,大幅降低研发成本,加速先导化合物的发现进程。根据可用的数据 (有无蛋白结构、活性化合物数量) 合理选择和组合这两种策略,是成功进行虚拟筛选的关键。
想让虚拟筛选又快又准?关键是“按需选择,合理组合”!
第一步:有蛋白结构→优先用 SBVS 精准对接;
第二步:有已知活性分子→先用 LBVS 快速初筛;
第三步:两者都有→用“LBVS 缩范围 +SBVS 提精度”的组合策略!
MCE 智能与药物设计平台,可综合运用分子对接、虚拟筛选、分子动力学模拟等技术,依托高性能服务器,提供基于配体/受体的 AI 筛选、分子动力学模拟、结构优化及化合物合成一体化服务——最短数小时内完成数千万分子筛选,真正实现快速高效,助力科研 er 破解药物筛选难题!
产品推荐
虚拟筛选 (Virtual Screening, VS) 是基于小分子数据库开展的活性化合物筛选。利用小分子化合物与药物靶标间的分子对接运算,虚拟筛选可快速从几十至上百万分子中,遴选出具有成药性的活性化合物,大大降低实验筛选化合物数量,缩短研究周期,降低药物研发的成本。
人工智能 (Artificial Intelligence,AI) 药物筛选是一种结合 AI 技术与计算化学的高通量筛选方法,广泛应用于蛋白结构预测、新药研发和分子设计与优化等领域。其主要目的是利用机器学习 (MachineLearning,ML) 算法分析大量数据,从中学习规律,生成 AI 打分函数,以此提高筛选效率,加速候选药物的发现过程。
分子对接 (Molecular Docking) 是一种基于计算模拟的药物分子与生物大分子 (如蛋白质、核酸等) 相互作用预测技术。其核心原理包括锁钥模型 (Lock-and-KeyModel) 和诱导契合学说 (InducedFitTheory) ,分别描述配体-受体的刚性匹配和动态构象调整过程。
分子动力学 (Molecular Dynamics, MD) 模拟是一种基于牛顿力学,综合了物理、数学和化学等多种学科的计算机模拟方法,用于研究分子体系的运动和相互作用、预测分子体系的行为和结构性质。通过计算机分子模拟,研究人员能够在分子水平上理解生物大分子的运动与生物功能、蛋白-小分子之间相互作用机理。
表面等离子共振 (Surface Plasmon Resonance,简称 SPR) 是借助传统光学现象,利用光在不同介质中产生消逝波后与等离子波产生共振,进而可以构建生物分子相互作用的生物传感分析技术,用以检测生物传感芯片上配位体与分析物之间的相互作用情况。
参考文献
[1]ZhangX, et al.Z-DNA-bindingprotein1exacerbatesmyocardialischemia‒reperfusioninjurybyinducingnoncanonicalcardiomyocytePANoptosis.SignalTransductTargetTher.2025Oct7; 10(1):333.
[2]GuttmanY,etal.DietaryInhibitorsofCYP3A4AreRevealedUsingVirtualScreeningbyUsingaNewDeep-LearningClassifier.JAgricFoodChem.2022Mar2; 70(8):2752-2761.
[3]ZuanonM,etal.IdentificationofNewHumanP2X7AntagonistsUsingLigand-andStructure-BasedVirtualScreening.JChemInfModel.2025Jul14; 65(13):7143-7155.

https://blog.sciencenet.cn/blog-3536222-1531964.html
上一篇:AACR 落幕,ai药物筛选、小分子药物、生物药、抗体偶联药物ADC 集亮相_MCE
下一篇:科研定制|MCE 实验动物饲料定制

全部作者的其他最新博文
- • HLX43 是一种靶向 PD-L1 的抗体偶联药物_ MedChemExpress (MCE)
- • PLX5622 in AIN-76A Diet (1200 ppm),1303420-67-8_ MCE
- • Trastuzumab,曲妥珠单抗,180288-69-1_ MedChemExpress (MCE)
- • Cell Counting Kit-8简称 CCK-8 试剂盒或 CCK8 _ MedChemExpress (MCE)
- • D-Luciferin potassium,D-荧光素钾然底物_ 盐_ MedChemExpress (MCE)
- • Phorbol 12-myristate 13-acetate,佛波酯,16561-29-8_ MCE