博文
EMD|洞见“锰”机——破译“死锰”密码,助力高能水系电池
||
第一作者:谢恺、朱鹏翰
通讯作者:韩大量、吴忠帅、翁哲
通讯单位:天津大学、中国科学院大连化学物理研究所
论文DOI:10.26599/EMD.2025.9370071
全文速览
基于双电子转移沉积/溶解化学的二氧化锰(MnO2),因其超高的理论比容量(616 mAh g−1)、高的氧化还原电位(相对于标准氢电极为1.23 V)、固有的无毒性和低成本特性,成为高能水系电池极具前景的正极候选材料。然而,其实际应用仍受制于电化学可逆性和循环稳定性差的问题,这主要归因于电化学惰性锰(Mn)物种(即“死锰”)的形成和积累。近日,天津大学Nanoyang团队的韩大量副研究员、翁哲教授与中国科学院大连化物所的吴忠帅研究员合作在Energy Materials and Devices发表了题为Decoding “Dead Mn” in MnO2 deposition/dissolution chemistry for energetic aqueous batteries: A perspective的观点论文,深入分析了Mn2+/MnO2化学中的“死锰”困境。首先,系统分析了“死锰”的根本原因——电子供应不足和质子供需失衡(不足或过量)——显著降低了MnO2的利用率、循环寿命和电池的能量密度。之后,从三个方面探讨了“死锰”问题的应对策略:(i) 防止因电子供应不足导致的“死锰”形成,(ii) 缓解质子供需失衡导致的“死锰”形成,以及 (iii) 激活和再利用已形成的“死锰”。最后,展望了未来的研究方向,以提升Mn2+/MnO2沉积/溶解化学的性能,推动高能水系电池的发展。
1 引言
可充电水系电池因其本征高安全性,在大规模储能应用中引起了广泛关注。特别是二氧化锰(MnO2)因其资源丰富、环境友好和成本低而被认为是一种极具前景的正极材料。自2012年水系锌离子电池(AZIBs)问世以来,基于单电子嵌入/脱出机制的MnO2正极被广泛研究。然而,基于固-固相变的嵌入型MnO2正极在反复充放电循环中不可避免地会存在结构坍塌和活性物质损失问题(例如,由Jahn-Teller效应引起的Mn物种溶解),这严重限制了其性能提升和实际应用。2018年,美国斯坦福大学的崔屹院士团队率先报道了基于液相Mn2+和固相MnO2之间双电子转移的沉积/溶解反应新路径,开发了高能水系H₂//MnO2电池。具体而言,充电时电解液中的Mn2+被氧化沉积为固相MnO2,放电时MnO2溶解为Mn2+(式1-2)。
沉积 (Mn2+ → MnO2): Mn2+ + 2H2O - 2e⁻ → MnO2 + 4H⁺ (式1)
溶解 (MnO2 → Mn2+): MnO2 + 4H⁺ + 2e⁻ → Mn2+ + 2H2O (式2)
Mn2+/MnO2沉积/溶解化学因其高理论比容量(616 mAh g⁻¹)和高氧化还原电位(1.23 V vs. SHE)而引起了广泛的研究兴趣。这些优势源于其双电子可逆转化,使得开发高比能量水系电池成为可能。2019年,澳大利亚阿德莱德大学的乔世璋院士团队首次报道了利用这种溶解/沉积机制的高电压(1.95 V)电解Zn//MnO₂电池。此后,通过探索替代的金属负极如铜(Cu)、铋(Bi)、锡(Sn)以及替代的非金属负极如硫(S),开发了多种体系。基于Mn2+/MnO2沉积/溶解化学的水系电池已成为大规模储能技术中极具竞争力的候选者。
尽管Mn2+/MnO2沉积/溶解化学具有巨大的应用潜力,但其实际应用仍受到循环稳定性差的严重阻碍。这一瓶颈在于充放电循环过程中电化学惰性含Mn物种的形成和积累,通常称为“死锰”。具体而言,“死锰”是指因Mn2+/MnO2沉积/溶解过程中还原不完全和/或副反应导致的惰性MnO2或其他含Mn化合物(例如MnOx、MnOOH、MxMnO2等,其中M代表阳离子),这些物质因失去电接触而失去电化学活性。本质上,这些“死锰”物种难以在后续充放电过程中继续被利用,因此无法贡献容量。针对Mn2+和MnO2的双电子沉积/溶解过程通常通过Mn(III)中间体介导的路径进行,而这些Mn(III)中间体的高效转化需要电子和质子的匹配供应。当这种供需不匹配时,Mn(III)物种就容易转化为“死锰”,“死锰”的持续积累极大地影响了电池的可逆性和长期稳定性。尽管研究人员已经逐渐认识到“死锰”的危害并进行了有益的探索来缓解它,但仍缺乏关于“死锰”形成机制的深入讨论以及有效缓解策略的全面综述。因此,全面理解“死锰”形成机制并制定有效的缓解策略对于提高基于Mn2+/MnO2化学的水系电池性能和推进其实际应用至关重要。
2.“死锰”的形成与危害
电化学惰性“死锰”的形成和积累是导致Mn2+/MnO2沉积/溶解反应可逆性差的主要因素。根据失效机制,我们将“死锰”分为两类:(1)电接触失效的Mn物种以及(2)反应不完全的Mn(III)产物。前者通常与正极失去电接触,使其无法有效参与后续的电化学反应。因此,它们不易被溶解并还原为活性Mn²⁺,从而无法贡献容量。后者通常与正极保持电接触,但由于质子供应不足,它们无法进一步被还原为Mn(II)物种。因此,本节详细阐述了Mn2+/MnO2化学中“死锰”的形成机制和危害。
2.1 “死锰”的成因
2.1.1 电子供应
MnO2与许多金属氧化物类似,通常表现出固有的低电子电导率(~10−5至~10−6S cm−1)。为了减轻沉积MnO2层内的电子传输阻碍,特别是防止其脱落或开裂,通常采用三维(3D)集流体,如碳布或碳毡,作为水系MnO2基电池的正极集流体,而不是传统的金属箔材(如Cu或Ti箔)。这些3D碳基集流体为MnO2沉积提供了更大的表面积和更多的电化学活性位点。然而,由于MnO2固有的低电导率,MnO2过厚沉积不可避免地阻碍了电极内的电子传输,导致MnO2的不均匀沉积和脱落(图1a)。不均匀的MnO2沉积通常导致电场分布不均,其中高电压下更倾向生成Mn3+并促进其随后的水解歧化反应(2Mn3+ + 2H2O → Mn2+ + MnO2 + 4H⁺)。具体而言,Mn3+的水解歧化可发生在任何与电解液接触的位置(例如,隔膜、负极、电池壳体等),导致MnO2在正极区域外形成,这种MnO2往往会失去电接触,变成“死锰”。
除了MnO2沉积外,放电过程中MnO2的电化学溶解也会导致“死锰”的形成。如式2所示,一个MnO2溶解需要四个质子,表明溶解是一个质子驱动的过程。通常,MnO2的外层首先发生电化学溶解,因为它最靠近提供质子的电解液。然而,当不均匀溶解或部分MnO2脱落暴露出集流体时,在三相边界(集流体/电解质/MnO2界面)的MnO2会发生加速溶解,而内层MnO2的溶解会导致外层MnO2颗粒失去电接触,从而形成中空的“死锰”壳。这个问题对于高面积容量下具有高内应力的厚MnO2沉积尤为显著。
2.1.2 质子供应
除了电子供应,电极/电解液界面的质子供应是“死锰”形成的另一主要原因。如式1和2所示,质子在MnO2沉积过程中生成,在溶解过程中被消耗,这使得整个过程对局部pH环境和质子可用性高度敏感。此外,MnO2的沉积/溶解通常通过Mn(II) → Mn(III) → Mn(IV)(或反向)的路径进行,由Mn(III)中间体介导,该过程显著受控于电极/电解液界面的质子供应速率,这直接决定了电化学反应能否发生。例如,我们前期工作证明Mn(III)中间体表现出强烈的pH依赖性:在强酸性电解质中,Mn3+占主导;而在近中性电解质中,倾向于形成羟基氧化锰(MnOOH)或其他低价氧化锰(MxMnO2)。在酸性环境(质子供应过量)中,高质子浓度促进Mn3+形成,加剧其歧化生成非电接触“死锰”。相反,在弱酸或者近中性电解液中(质子供应不足),有限的质子供应阻碍了MnO2的进一步溶解,导致导电性差的MnOOH或MxMnO2而非目标产物Mn2+的积累(图1a)。总之,维持适中的质子供应对于Mn2+/MnO2沉积/溶解化学至关重要:过量的质子供应促进Mn3+形成,而不足的质子供应阻碍MnO2的完全溶解,导致低电导率Mn(III)化合物的积累。
然而,在Mn2+/MnO2化学中,易歧化的Mn3+和低电导率的固态Mn(III)化合物(例如,MnOOH、MxMnO2等)均非目标产物(Mn²⁺或MnO2)。在高局部质子浓度(pH < 2)下通常会形成可溶的Mn3+物种,其容易从电极表面扩散离开并在非电接触区域发生水解歧化反应(2Mn3+ + 2H2O → Mn2+ + MnO2 + 4H⁺),使得这部分Mn物种在后续循环中失去电化学活性。与高质子浓度相比,在质子供应不足时会导致的固态Mn(III)中间产物积累,其电导率比MnO2更低,因此会导致电极表面钝化并阻碍后续的电化学过程。如上所述,电极/电解液界面质子供应过多或过少,都会直接导致“死锰”的形成,从而降低了体系的可逆性。
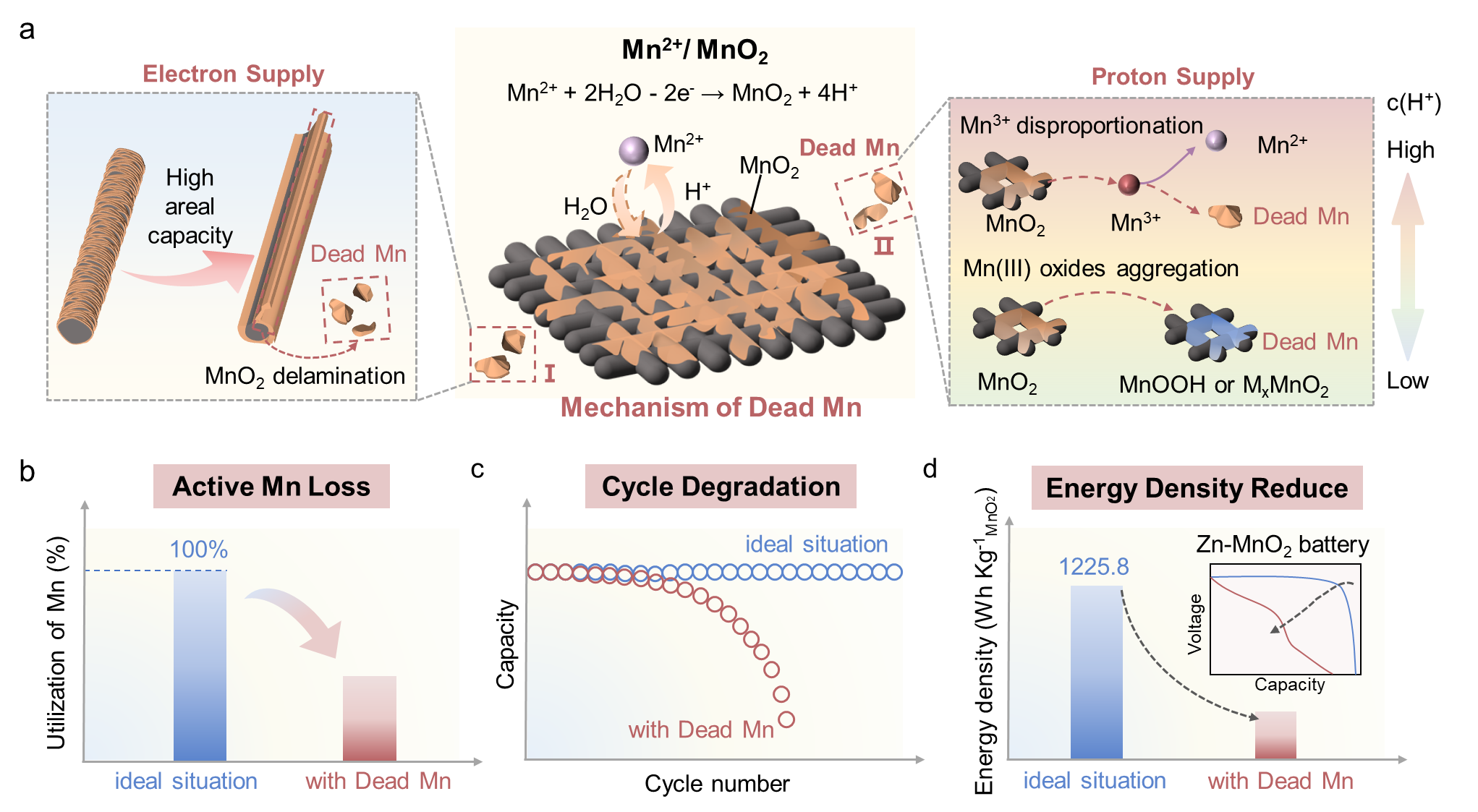
图1 “死锰”的形成机制和危害
2.2 “死锰”的危害
电化学惰性“死锰”的不可避免形成和积累是导致Mn2+/MnO2沉积/溶解化学可逆性差的主要原因。“死锰”的危害主要体现在三个方面:(i)活性Mn物种的不可逆损失,(ii)容量快速衰减,以及(iii)能量密度降低(图1b–d)。“死锰”最直接的影响是活性Mn物种的不可逆损失:通过Mn3+歧化、MnO2脱落或形成某些低电导率锰氧化物产生的电化学惰性“死锰”,无法或难以参与后续的充放电循环,从而导致活性物质逐渐减少。因此,电池会经历快速的容量衰减,同时循环稳定性变差。“死锰”的积累也加剧了电极钝化,同时阻塞了离子传输路径,导致极化增加和实际能量密度低(远低于理论值;图1d)。“死锰”的上述危害极大地限制了水系MnO2基电池在大规模储能应用中的竞争力,突显了制定高效缓解策略的迫切需求。
3 “死锰”的缓解策略
针对“死锰”挑战,已报道了几种有效的策略。我们首次从电子供应和质子供应的角度系统地对现有缓解策略进行了分类,将其分为三种不同的方法:(i)通过基底设计、结构控制和充电程序(协议)优化来优化沉积过程电子供应来防止“死锰”形成;(ii)通过调节界面反应环境和反应路径调控质子供应来缓解“死锰”形成;(iii)通过使用氧化还原介体(RMs)等方法激活和再利用已形成的“死锰”物种。
3.1 优化电子供应
MnO2本征的低电子电导率导致电子传输困难,这在高负载条件下尤其容易导致电接触失效。对于常见的嵌入型MnO2正极,导电性的提升通常通过晶格掺杂和表面包覆等方法实现,而上述改性通常是在材料合成时进行的。而基于嵌入机制的MnO2理想情况下几乎不发生溶解和沉积,因此上述增强导电性的策略可以在整个循环中持续发挥作用。相比之下,上述改性策略对于沉积/溶解型MnO2正极仍面临严峻挑战:因为基于该机制的活性物质MnO2在每个循环中都是“动态变化”的,其在放电过程中溶解并在充电过程中重新沉积,因此就要求改性策略在每个循环中都能保持有效。然而,通常在材料合成阶段实施的改性方案很难适用于电沉积过程,因此很难通过直接简单借鉴嵌入型MnO2的改性策略来提高沉积/溶解型MnO2的导电性。为解决“死锰”问题和提高Mn2+/MnO2可逆性,研究人员探索了各种替代方法,包括增强导电性(阳离子共沉积构建结构缺陷、调控沉积温度改善沉积晶型等)、优化沉积动力学(调控Mn2+溶剂化结构高效沉积等)、改性基底(增加反应活性位点优化离子和电子传输路径等)以及优化充电协议(调控沉积速率、电位等条件)。这些方法旨在从一开始就避免“死锰”物种的形成,从而减少因电接触失效引起的“死锰”积累,并使得开发高可逆、高容量的水系MnO2基电池成为可能,充分发挥其固有的高能量密度优势。
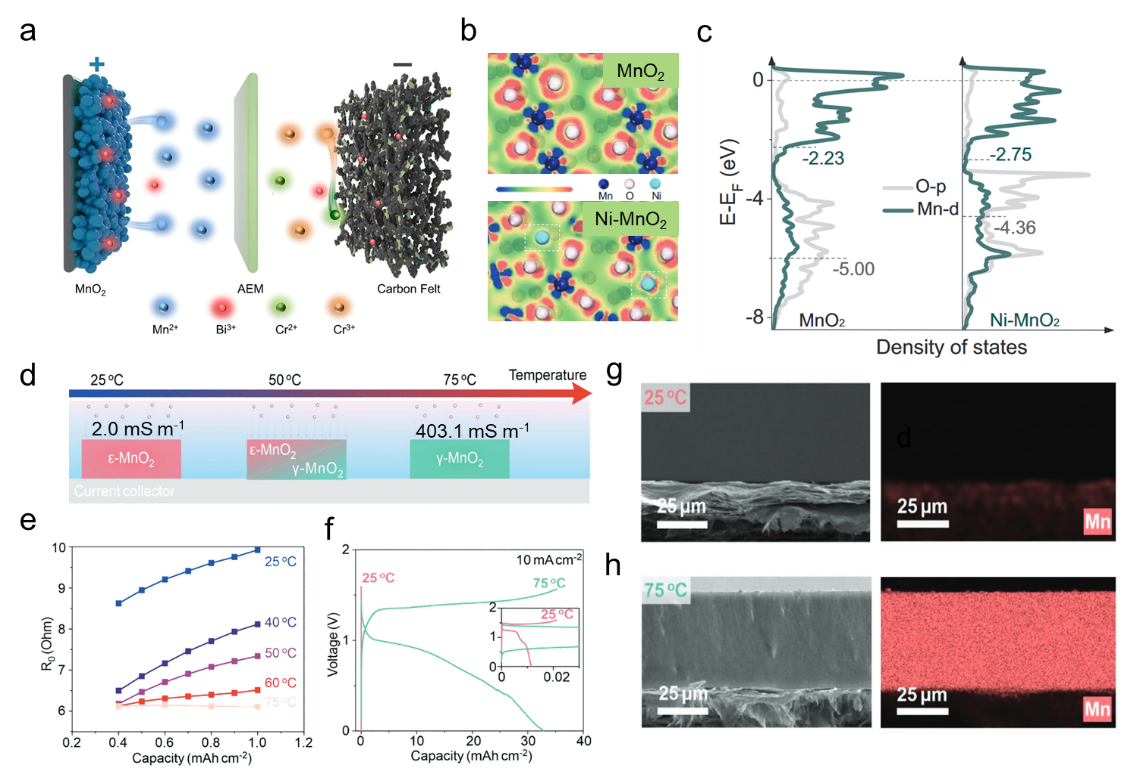
图2 (a) MnO2//Cr(Bi)电池示意图。(b) MnO2和Ni-MnO2的电子密度差俯视图。(c) MnO2和Ni- MnO2中O p带(灰色)和Mn d带(绿色)的部分态密度(PDOS)及能带中心值。(d) 沉积的MnO2晶型随温度变化的示意图。(e) 不同温度下MnO2沉积容量对应的电池电阻。(f) 不同温度下MnO2//H2电池的充放电曲线。(g, h) 石墨箔上沉积MnO2层的横截面SEM图像及对应的能量色散X射线谱(EDS)图谱。
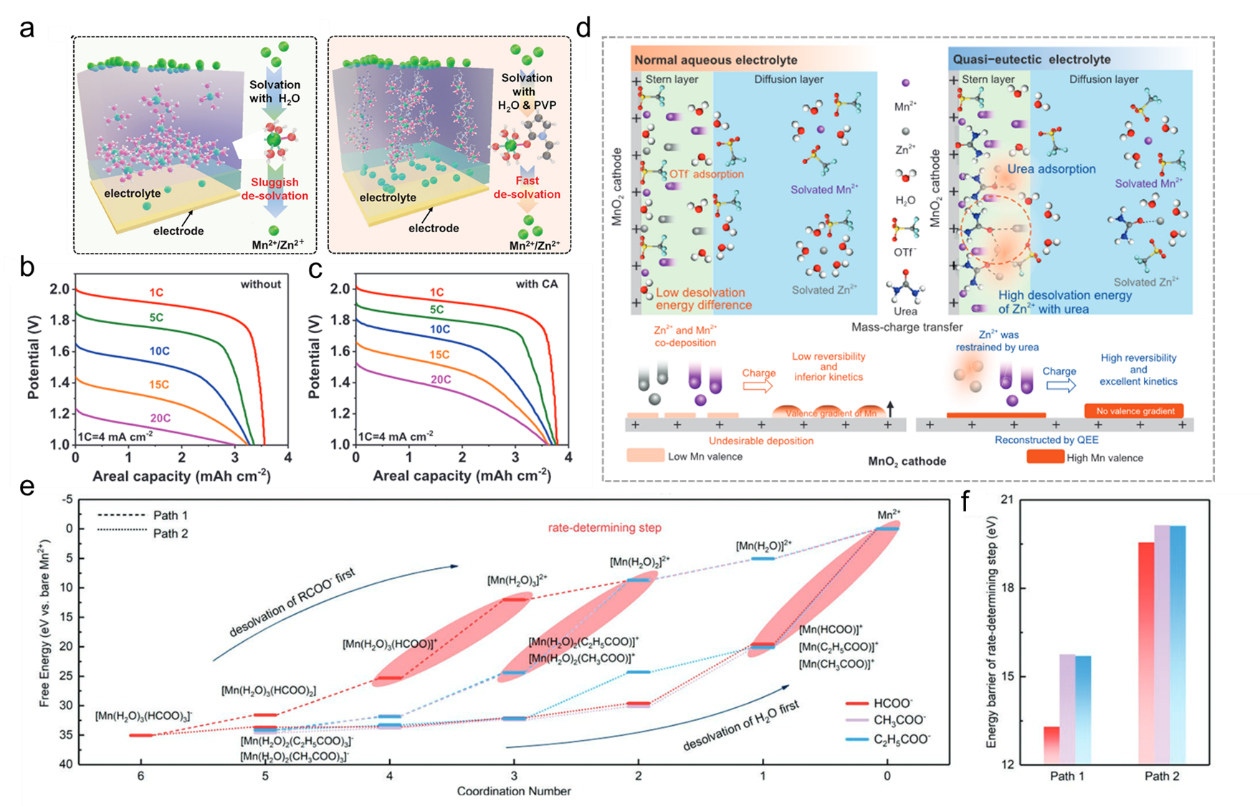
图3 (a) 不同电解液中Mn2+沉积过程的示意图。(b, c) Zn//MnO2电池在1至20 C倍率下的放电曲线:(b) 不含CA,(c) 含CA。(d) 常规电解液与添加尿素的电解液中正极界面的示意图。(e) Mn2+脱溶剂化反应路径及(f) 决速步骤的能垒。
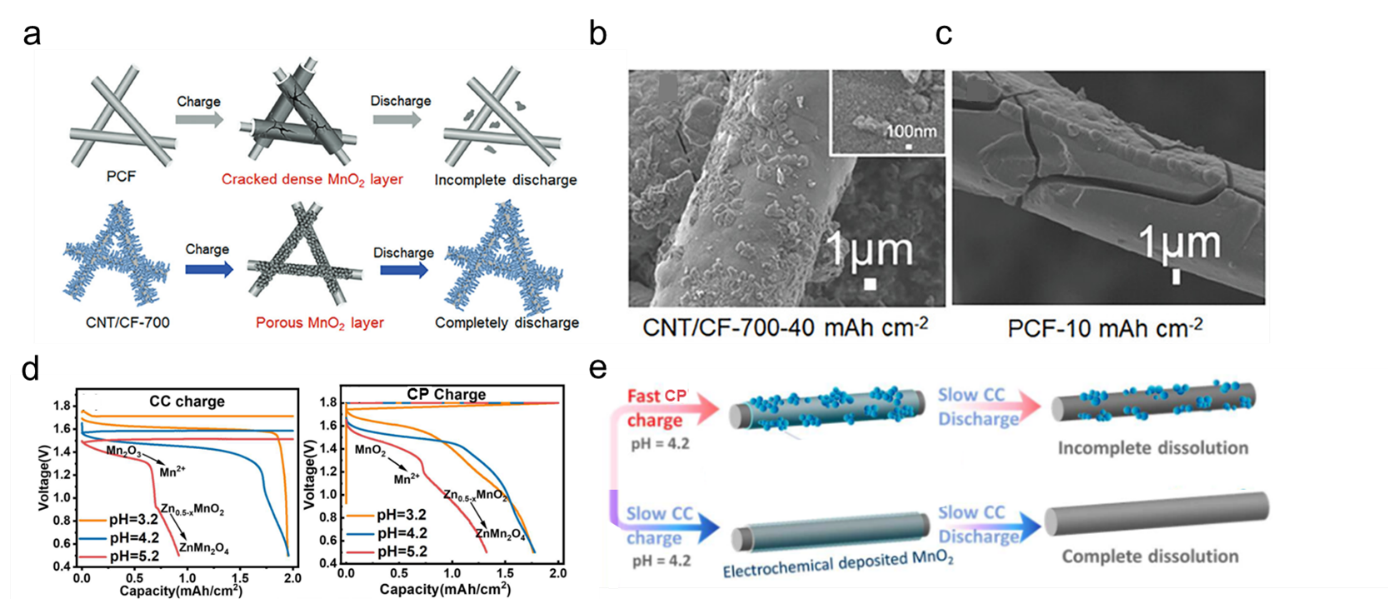
图4 (a) PCF和CNT/CF-700基底用于MnO2沉积的示意图。(b) CNT/CF-700电极上沉积40 mAh cm−2MnO2的SEM图像。(c) PCF上沉积10 mAh cm−2MnO2₂的SEM图像。(d) 不同充电协议下的充放电曲线。(e) 不同条件下MnO2沉积的示意图。
3.2 调控质子供应
Mn(III)中间体的存在形式和稳定性与界面质子供应密切相关,其直接影响MnO2沉积/溶解过程的可逆性。特别是,在高质子浓度的酸性环境中,热力学上有利于Mn3+形成,加剧歧化反应,导致电化学惰性“死锰”的积累。相反,在低质子浓度的近中性环境中,有限的质子阻碍了MnO2进一步溶解,导致低活性Mn(III)化合物(如MnOOH)的积累,这些化合物的电子和离子电导率低,其电化学活性差。因此,抑制Mn(III)中间体形成和调控电解液质子供应是缓解“死锰”的关键。
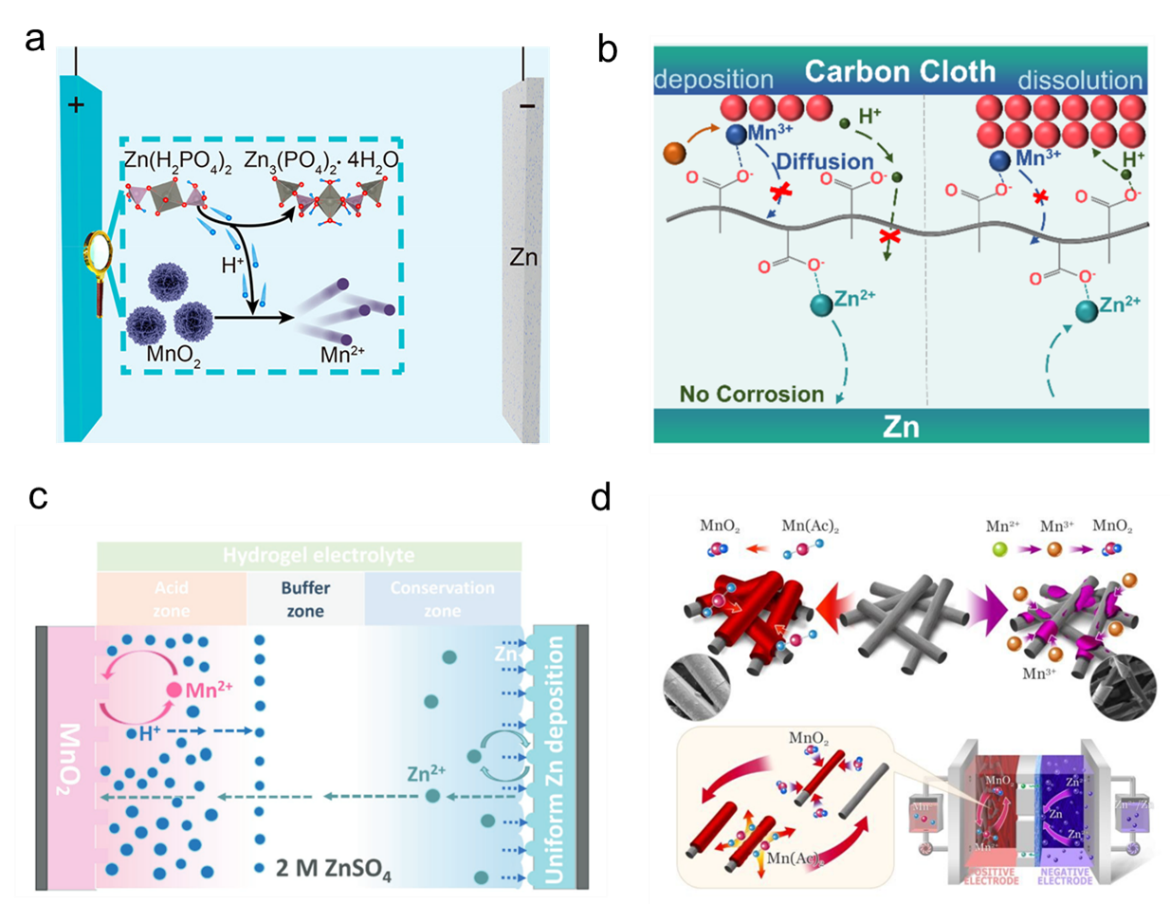
图5 (a) Zn(H2PO4)2调控Mn2+/MnO2沉积/溶解的工作机制示意图。(b) 原位水凝胶离子锚定策略限制Mn3+和质子扩散的示意图。(c) 三层水凝胶电解质Zn//MnO2电池示意图。(d) 乙酸锰(Mn(Ac)2)电解质中Mn2+/MnO2沉积/溶解的示意图。
3.3 激活和再利用已生成的“死锰”
尽管优化沉积条件和调控界面质子供应已被证明有效,但完全防止“死锰”的形成在根本上仍然具有挑战性。为了实现高可逆性,需要活化和再利用已形成的“死锰”,以恢复其电化学活性并提高活性材料的利用率。氧化还原介质(RMs)在重新激活“死锰”方面显示出巨大的应用潜力,它们利用其可逆的氧化还原循环来促进电荷转移,而不改变最终反应产物。其潜在机制依赖于RMs在氧化态和还原态之间可逆转换的能力,使其能够通过提供额外的电荷转移途径来调节反应动力学。在MnO2沉积/溶解过程中,RMs作为还原剂将“死MnO2”电化学还原,以恢复其电化学活性。为了有效促进这一过程,RMs的氧化还原电位必须低于Mn2+/MnO2电对的氧化还原电位(图6a)。迄今为止,几种可溶且成本效益高的RMs,如Fe2+/Fe3+, I⁻/I₃⁻, Br⁻/Br₃⁻, VO²⁺/VO₂⁺和1,4-苯醌(1,4-BQ)/氢醌(HQ),均已被探索用于再生“死MnO2”,并在增强MnO2基水系电池的循环寿命和库伦效率方面显示出良好效果。
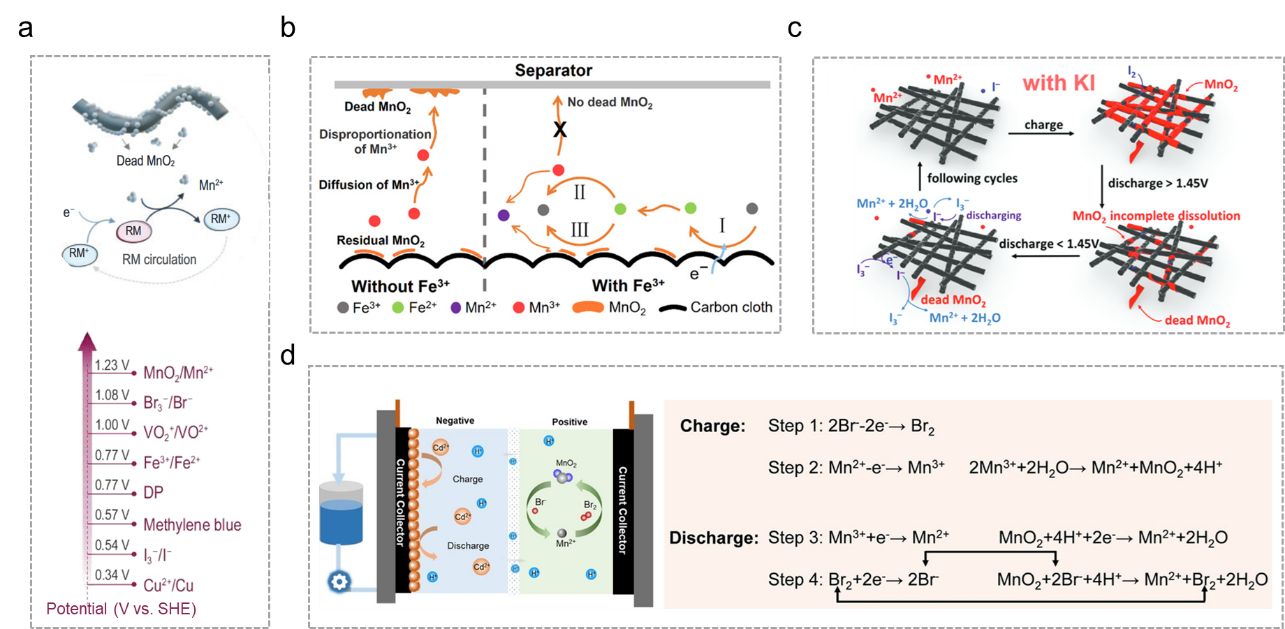
图6 (a) 消除“死MnO2”的氧化还原介质(RM)设计及不同氧化还原电对的电位。(b) “死MnO2”机制及Fe2+/Fe3+RM的作用机理示意图。(c) I⁻/I₂ RM解决“死MnO2”问题的示意图。(d) Br⁻活化“死MnO₂”的示意图及电化学机制总结。
4 结论与展望
对于基于Mn2+/MnO2沉积/溶解化学的水系电池,其在大规模储能中的广泛应用目前受到电化学可逆性差和循环稳定性有限的阻碍。这些限制主要归因于电化学惰性“死锰”的形成和积累。本观点论文剖析了“死锰”形成的根本原因,明确了电子供应不足和质子供需不匹配是关键的机制。相应地,系统地综述了针对“死锰”形成和积累的缓解策略,包括:(i) 优化MnO2沉积以改善电子传输,缓解电子供应不足;(ii) 调控质子供应以减少Mn(III)中间体相关的“死锰”形成;(iii) 通过氧化还原介体(RMs)等方法激活和再利用已形成的“死锰”以恢复其电化学活性。对基本挑战和潜在解决方案的系统分析,为解决“死锰”问题和开发高性能水系MnO2基电池提供了理论指导。
此外,通过优化质子和电子的供应对于防止“死锰”的形成,增强Mn2+/MnO2化学的电化学可逆性至关重要。在提升基于Mn2+/MnO2沉积/溶解化学的水系电池的实际可行性方面,未来的研究应聚焦于以下四个关键领域(图7):
1)进一步优化MnO2材料的本征结构:MnO2本征物理化学性质直接影响其电化学活性、离子/电子传输效率以及循环过程中的结构稳定性。因此,开发具有高导电性和电化学活性的MnO2是实现电子-质子匹配、构建高可逆水系MnO2基电池的关键。具体策略包括:(i) 调控沉积MnO2的形态工程(如纳米棒、球形颗粒或堆叠纳米片)以优化电场分布和与电解质的良好接触;(ii) 通过阳离子共沉积在MnO2中引入结构缺陷(如空位),确保持续的缺陷介导的导电性和电化学活性增强贯穿动态沉积/溶解过程;(iii) 控制从无定形ε-MnO2向高导电性稳定MnO2相的相变。这些策略可增强导电性,减少与“死锰”形成相关的电化学接触失效,并提高电化学可逆性和循环稳定性。
2)电解液设计与定制:作为离子传输和电化学反应的介质,电解液的组成和性质在Mn2+/MnO2沉积/溶解行为中起着决定性作用。然而,沉积和溶解所需的界面微环境差异很大。例如,它们对质子需求是矛盾的,目前还没有电解液能精确满足这些反应要求。因此,未来研究应侧重于设计和定制电解质液以满足特定的界面反应需求,从而实现高度可逆的沉积/溶解过程。潜在方法包括:(i) 精细调整电解液pH值以精确匹配电化学反应要求,探索最佳pH范围或开发pH自适应调节系统,以减轻强酸性环境对负极和电池辅助部件的不利影响;(ii) 通过阴离子选择(如SO₄²⁻、Ac⁻、Cl⁻、TFSI⁻)、调整电解质盐浓度(如“盐包水”电解质)或使用混合溶剂系统来控制溶剂化结构,建立合理的溶剂化原则;(iii) 开发有效的电解液添加剂,如表面活性剂和RMs,以促进均匀的MnO2沉积和完全溶解,旨在增强可逆性。
3)开发新的评估指标以准确评估性能:准确评估电化学可逆性并深入理解电化学储能器件失效机制对于优化其性能是必要的。鉴于复杂的双电子Mn2+/MnO2沉积/溶解机制,可能发生多种副反应和失效模式,使得常规评估指标(如库伦效率)存在局限性。因此,建立更准确的评估方法,例如能够量化“死锰”比例的方法,对于有效指导性能优化非常重要。开发这些新指标需要全面理解潜在的储能机制,这可能需要界面表征技术来监测界面电化学反应。例如,监测沉积反应动力学可以防止“死锰”的不均匀沉积。此外,开发高精度界面pH监测方法可最大程度减少溶解过程中“死锰”的形成。再者,原位电化学石英晶体微天平(EQCM)可用于解耦和分析伴随沉积/溶解反应的电子转移过程。
4)提高器件级能量密度:能量密度是决定储能技术经济可行性和实际适用性的关键参数。在确保高可逆性、稳定循环和优异安全性的同时,提升基于Mn2+/MnO2水系电池的能量密度,需要在正极、负极和器件层面取得进步。提高能量密度的策略包括:(i) 提升MnO2面载量以构建厚电极;(ii) 开发低电位金属负极(如Zn)的兼容性策略,以构建高工作电压全电池;(iii) 优化解耦正极/负极电池器件的成本和结构,以最小化电解液用量从而提高能量密度,同时降低离子交换膜和其他辅助材料的成本,从而有助于开发高能量密度和低成本的器件。
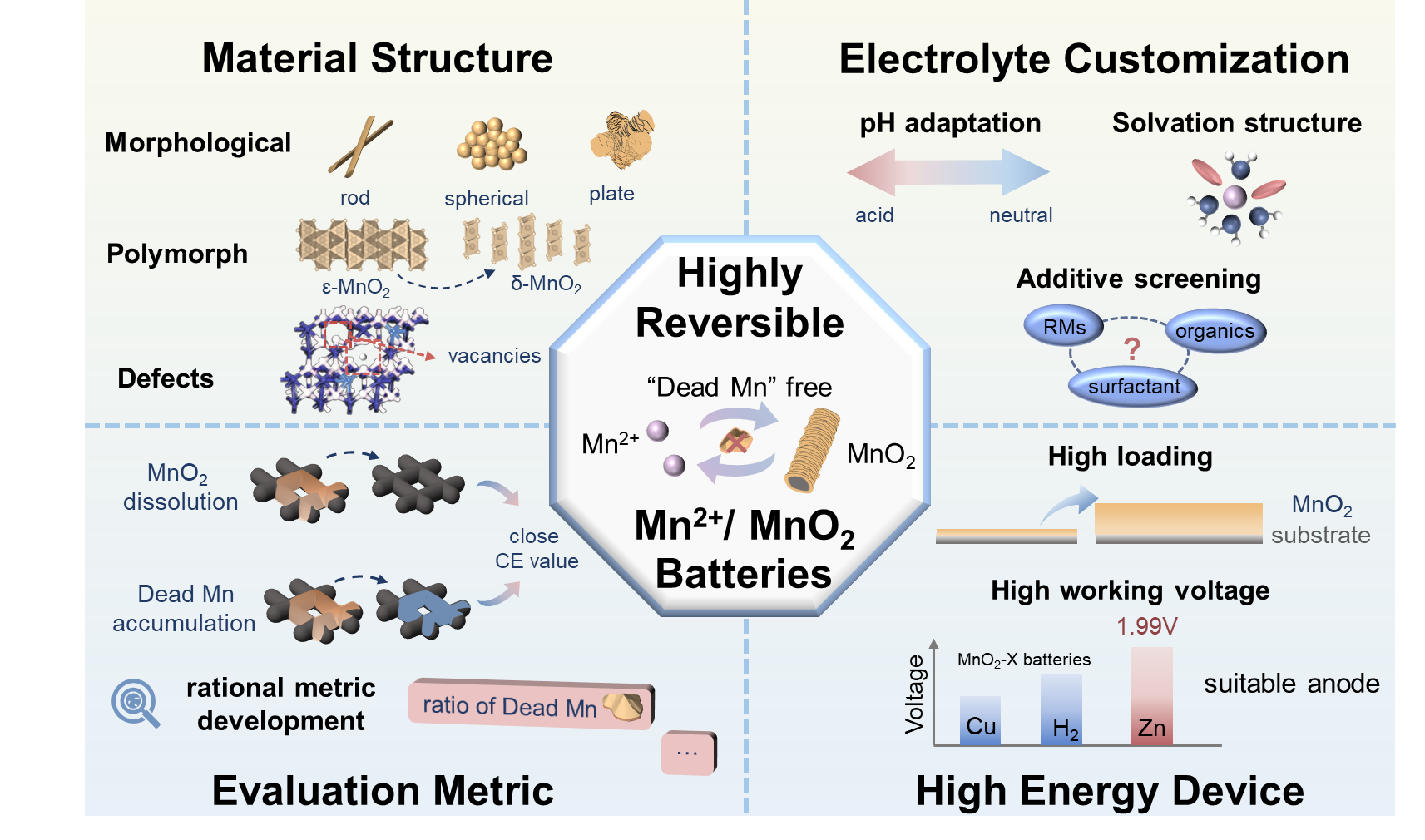
图7. 基于Mn²⁺/MnO₂化学的高能水系电池的未来研究方向
通讯作者:
翁哲,天津大学化工学院,英才教授,博士生导师。主要从事水系锌离子电池、电催化二氧化碳还原等方向的研究,在Nat Sustain, Nat Commun, J Am Chem Soc, Angew Chem Int Ed, Adv Mater, Energy Environ Sci等国际期刊发表研究论文60余篇,被他引8600余次,授权发明专利7件,参与制定水系金属离子电池团体标准2项;获2024年天津市自然科学一等奖1项(排名第三)、2024年中国青年科技奖天津市提名;主持国家重点研发计划课题、国家自然科学基金面上项目、天津市自然基金面上项目等多项省/部级项目;任电化学、Renewables、Materials Reports Energy、Green Carbon等期刊青年编委。
吴忠帅,中国科学院大连化学物理研究所,首席研究员,博士生导师,二维材料化学与能源应用研究组(508组)组长,国家杰出青年科学基金获得者,英国皇家化学会会士。2018-2024年连续七年科睿唯安全球高被引科学家、爱思唯尔“中国高被引学者”、超级电容产业联盟青年工作委员会副主任。主要从事二维材料化学与微纳电化学能源应用的基础研究。已在Nature、Nat. Commun.(6篇)、Energy Environ. Sci.(16篇)、Adv. Mater.(20篇)、J. Am. Chem. Soc.(9篇)、Angew. Chem. Int. Ed.(12篇)、Natl. Sci. Rev.(9篇)等期刊发表学术论文360余篇,论文被引47000余次,H-index为99。申请发明专利180余项。承担中组部、科技部、基金委、省市地方以及企业横向项目等30余项。获批国际标准1项、国家标准2项。曾获辽宁省自然科学一等奖(2022,排名第一;2015,排名第四)、国家自然科学二等奖(2017,排名第四)、第十三届辽宁青年科技奖(2021)、Nano Research新锐青年科学家奖、Energy Storage Materials青年科学家奖、中国科学院优秀导师奖、卢嘉锡优秀导师奖、中国科学院大学领雁银奖-振翅奖等,2018-2023年连续六年入选“科睿唯安”全球高被引科学家,入选爱思唯尔“中国高被引学者”。兼任Energy Storage Materials、Applied Surface Science副主编,J. Energy Chem.执行编委,Natl Sci. Rev.编辑工作组成员,Science Bulletin、科学通报、Nanomaterials、Carbon Futures、Mater. Res. Express、Physics编委,Interdisciplinary Materials学术编辑,Chin. Chem. Lett.、eScience、Materials Futures、物理化学学报青年编委、Engineering通讯专家。
韩大量,天津大学化工学院副研究员,主要从事高安全电化学能源存储与转化相关研究。以第一/通讯(含共同)作者在Nat. Sustain.、Nat. Commun.、Adv. Mater.、Angew. Chem. Int. Ed.、Energy Environ. Sci.、Adv. Energy Mater.、Chem、Energy Storage Mater.等高水平期刊发表论文20余篇,被引3800余次;获天津市自然科学一等奖、Nano Research Energy学术新星金奖等荣誉;主持国家自然科学基金面上和青年项目、国家重点研发计划子课题、中国博士后科学基金特别资助项目和面上项目、天津市自然科学基金等多项基金;担任eScience、Energy Materials and Devices、Carbon Neutralization等期刊青年编委。
原文链接:https://www.sciopen.com/article/10.26599/EMD.2025.9370071


https://blog.sciencenet.cn/blog-3534092-1527086.html
上一篇:武汉纺织大学杜康、魏国超和汪胜祥等:枪晶石型Ca3SnSi2O9基陶瓷超高Q×f值的设计新范式与应用研究
下一篇:西南交通大学联合国内外十一所高校发表新综述:多场耦合烧结技术引领高性能材料制备新纪元
