博文
Cell Metabolism:内皮焕新,代谢长青
||
代谢学人
Cell Metabolism:内皮焕新,代谢长青
初稿 | 李姿萱
校对 | 周文豪
小编注 | 陈明洁 皮婉莹 郭钰涵
排版 | 陈明洁
编辑 | 孟美瑶

背景介绍
代谢稳态的维持依赖于动态且多向的器官间通讯。既往的研究大多集中于主要代谢组织(即肝脏、骨骼肌、脂肪组织、胰腺和大脑)之间的相互作用,并且已被证实器官间通讯的失调会增加肥胖和2型糖尿病(T2D)的发病风险。
内皮细胞(ECs)位于心血管系统的管腔内表面,参与止血、炎症、血管生成、血液与组织间物质交换等诸多生物学过程,分布广泛,数量庞大且表面积巨大,这些独特的功能和解剖学特征使内皮细胞能够同时监测全身和局部的信号,整合这些信息并做出适当的反应。由于内皮细胞在调节营养供需中发挥着关键作用,它们常被称为机体的“代谢守门人”。此外,内皮细胞功能障碍是代谢病理学和衰老的重要标志。然而,内皮细胞作为全身代谢的潜在贡献者,受到的关注相对较少。
线粒体是调节细胞能量代谢的关键细胞器,协调信号传导与效应机制以适应不断变化的代谢环境和需求。线粒体动力学是维持线粒体结构和功能稳态的关键动态过程,这种过程能够调节线粒体的分布、与其他细胞器相互作用,并通过分裂和融合形成线粒体网络。线粒体融合主要由动力蛋白相关GTP酶——线粒体融合蛋白MFN1和MFN2介导。从生理学角度看,线粒体动力学是一种确保细胞在应激条件下生存并维持稳态的基本适应机制。这与“线粒体低毒兴奋效应”(Mitohormesis)的概念相一致(小编注:线粒体低毒兴奋效应(Mitohormesis) 指细胞线粒体在受到轻度应激刺激时,会触发一系列适应性反应,从而增强细胞的抗逆能力)。
既往的研究中,营养条件的变化与线粒体形态之间的相互作用已被证实(小编注:营养过剩、严重应激或氧化磷酸化功能障碍会导致线粒体碎片化。而营养剥夺、轻度应激和氧化磷酸化增强则导致线粒体融合增加。参考文献:Sprenger HG, Langer T. The Good and the Bad of Mitochondrial Breakups. Trends Cell Biol. 2019;29(11):888-900.),由于内皮细胞主要依靠糖酵解产生ATP,线粒体在内皮细胞中的作用一直被忽视。近期研究表明,线粒体呼吸和功能对于血管生成至关重要。药物抑制内皮细胞中的线粒体呼吸链复合体III会破坏NAD+/NADH比例,导致内皮细胞增殖减少,并损害视网膜和肿瘤血管生成(参考文献:Diebold LP, Gil HJ, Gao P, Martinez CA, Weinberg SE, Chandel NS. Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat Metab. 2019;1(1):158-171.)。氧化磷酸化是内皮细胞代谢中的重要参与者,有研究人员通过敲除内皮细胞中的氧化磷酸化关键基因(细胞色素c氧化酶(COX)),构建内皮细胞氧化磷酸化缺失的小鼠,结果发现氧化磷酸化缺失会导致血管发育缺陷和功能障碍,导致胚胎致死、伤口愈合延迟,同时也能导致肺癌和黑色素皮下瘤小鼠的血管生成减少,延缓肿瘤发展进程(参考文献:Schiffmann LM, Werthenbach JP, Heintges-Kleinhofer F, et al. Mitochondrial respiration controls neoangiogenesis during wound healing and tumour growth. Nat Commun. 2020;11(1):3653.)。此外,线粒体内膜融合蛋白视神经萎缩蛋白1(OPA1)对血管生成至关重要,在血管生成因子的刺激下,OPA1水平会迅速升高,从而抑制NFkB信号传导,最终促进血管生成相关基因的表达及血管生成(参考文献:Herkenne S, Ek O, Zamberlan M, et al. Developmental and Tumor Angiogenesis Requires the Mitochondria-Shaping Protein Opa1. Cell Metab. 2020;31(5):987-1003.e8.)。然而,内皮细胞线粒体在除生物能量学之外发挥的作用尚未被深入研究。
近期,一篇发表在《Cell Metabolism》上的文章证明了线粒体动力学在内皮细胞(ECs)代谢中的作用。研究人员发现,饮食诱导肥胖小鼠白色脂肪组织中的内皮细胞存在线粒体融合缺陷。然而,在成年小鼠的ECs中敲除Mfn2会触发线粒体低毒兴奋效应(Mitohormesis),进而改善全身代谢和衰老表型。总之,该研究揭示了内皮细胞中Mfn2与线粒体低毒兴奋效应之间的联系,这种联系有助于维持全身代谢和机体健康。

敲黑板啦
1.内皮细胞中MFN2的缺失触发白色脂肪线粒体低毒兴奋效应
2.内皮细胞MFN2的缺失增强小鼠的脂质氧化并使其脂肪量降低
3.内皮细胞通过FOXO1分泌GDF15,MFN2缺失会升高GDF15表达水平
4.内皮细胞缺乏MFN2的小鼠能够抵御肥胖和衰老相关的功能衰退
研究结果
1、代谢紊乱影响小鼠白色脂肪组织内皮细胞的线粒体动力学
为了研究病理状态下内皮细胞的线粒体动力学,研究人员选择了特别容易受肥胖影响的白色脂肪组织内皮细胞,研究人员分析了饮食诱导肥胖(HFD)小鼠和对照小鼠的脂肪内皮细胞线粒体。结果发现,与对照小鼠相比,HFD小鼠的eWAT中线粒体长宽比、面积、覆盖率和密度均降低,表明其线粒体融合减少(图1A–1E)。MFN是线粒体融合的关键调节因子。因此,研究人员构建了Pdgfb-iCreER:Rpl22(RibotagiEC)小鼠(小编注:Ribotag小鼠的Rpl22(核糖体蛋白)基因序列上插入了loxp位点和HA(血凝素A)位点,当Ribotag小鼠与Pdgfb-iCreERT2小鼠杂交后,Pdgfb启动子驱动他莫昔芬诱导型Cre重组酶(iCreER)在内皮细胞中特异性表达。这里在小鼠8-12周龄时,通过连续五天每天腹腔注射一次他莫昔芬。Cre-ERT2小鼠是一类含有雌激素受体(ER)的配体结合区突变体(ERT,突变是为了消除内源性雌激素的干扰)与Cre重组酶的融合蛋白表达的小鼠。Cre-ERT2在无他莫昔芬诱导的情况下,在细胞质内处于无活性状态;当他莫昔芬诱导后,他莫昔芬的代谢产物4-OHT(雌激素类似物)与ERT结合,可使Cre-ERT2进核发挥Cre重组酶活性。激活后的Cre重组酶可介导Rpl22-HA等位基因的重组,使HA标签序列稳定整合至核糖体蛋白Rpl22上,由此,在内皮细胞中,所有活跃翻译的核糖体均被HA标记。使用针对HA的单克隆抗体对多聚核糖体进行免疫沉淀,可从特定细胞类型中分离出核糖体结合的mRNA转录本。(参考文献:Sanz E, Yang L, Su T, Morris DR, McKnight GS, Amieux PS. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci U S A. 2009;106(33):13939-13944.))以检测肥胖条件下白色脂肪组织内皮细胞中Mfn1和Mfn2的表达情况。为了验证该小鼠模型,研究人员通过HA标签免疫荧光染色,确认了Rpl22融合蛋白在内皮细胞中的特异性表达;然后检测到核糖体免疫沉淀产物中内皮细胞标志物——血管内皮钙粘蛋白5(Cdh5)的富集,验证了该模型构建成功(图S1A和S1B)。RibotagiEC小鼠接受标准饲料或高脂饮食(HFD)喂养12周后(图S1C),HFD小鼠出现明显的肥胖和空腹高血糖(图S1D–S1F)。与线粒体形态学数据一致,与对照组相比,HFD小鼠WAT的内皮细胞中Mfn1和Mfn2的表达显著降低(图1F和1G)。这些结果表明,肥胖会减少WAT中内皮细胞的线粒体融合。
2、内皮细胞缺失Mfn2会改善机体代谢,增强脂质动员
首先,研究人员研究了Mfn1在内皮细胞中的功能。研究人员将Mfn1fl/fl小鼠与Pdgfb-iCreERT2小鼠杂交,获得了内皮细胞特异性Mfn1敲除小鼠(Mfn1iΔEC),并通过琼脂糖电泳验证了Cre介导的重组发生在大多数组织中(图S2A)。在普通饮食和高脂饮食条件下,对照小鼠与Mfn1iΔEC小鼠的体重和葡萄糖代谢均没有差异(图S2B–S2H)。接下来,研究人员构建了内皮细胞特异性Mfn2敲除小鼠(Mfn2iΔEC),发现Cre介导的重组发生在大多数成年组织中(此处是指成年小鼠的组织样本)(图S2I),并未引起组织病理学的改变(表S1)。相比于对照小鼠,雄性Mfn2 iΔEC小鼠的体重增长速度明显减慢,5周后对照小鼠和Mfn2 iΔEC小鼠的体重差异趋于稳定(Mfn2 iΔEC小鼠约降低20%)(图1H)。体成分分析显示,Mfn2 iΔEC小鼠脂肪量显著降低,而肌肉质量与对照小鼠没有差异(图1I)。一致地,Mfn2 iΔEC小鼠腹股沟脂肪组织(iWAT)和附睾脂肪组织(eWAT)质量以及脂肪细胞尺寸显著减小,而肩胛间棕色脂肪组织(BAT)质量没有变化(图1J,1K,S2J和S2K)。从Mfn2iΔEC小鼠分离的基质血管组分细胞能够正常分化为脂肪细胞,并表达相同水平的Mfn2和经典的脂肪细胞分化标志基因(图S2L–S2S)。此外,Mfn2iΔEC小鼠的葡萄糖代谢稳态得到改善(图1L和1M),iWAT中胰岛素信号传导增强(图S2U),葡萄糖刺激的胰岛素分泌保持不变(图S2V)。雌性Mfn2iΔEC小鼠的体重和脂肪重量也呈现出与雄性小鼠相似的表型(图S2W和S2X)。
与体重下降相一致的是,雄性Mfn2iΔEC小鼠在12-13周龄时的平均食物摄入量略有减少(图1N),营养吸收能力、活动量和能量消耗均无差异(图S3A–S3F),产热脂肪的活性、产热相关基因表达均无差异(图S3G–S3J)。然而,Mfn2iΔEC小鼠的呼吸熵(RQ=VCO2/VO2)呈降低趋势,尤其在夜间阶段更为明显,表明脂肪氧化增强(图1O和S3K)。此外,与对照小鼠相比,Mfn2iΔEC小鼠的激素敏感性脂肪酶磷酸化水平升高,说明脂解作用增强(图1P和S3L)。禁食状态下血浆游离脂肪酸水平增加(图1Q),而脂质摄取与合成相关基因的表达与对照小鼠相比没有变化(图S3M)。这些结果表明,Mfn2iΔEC小鼠体重和脂肪量减少的原因,可能是食物摄入量轻微减少,以及白色脂肪组织脂解作用和脂肪酸氧化增强。
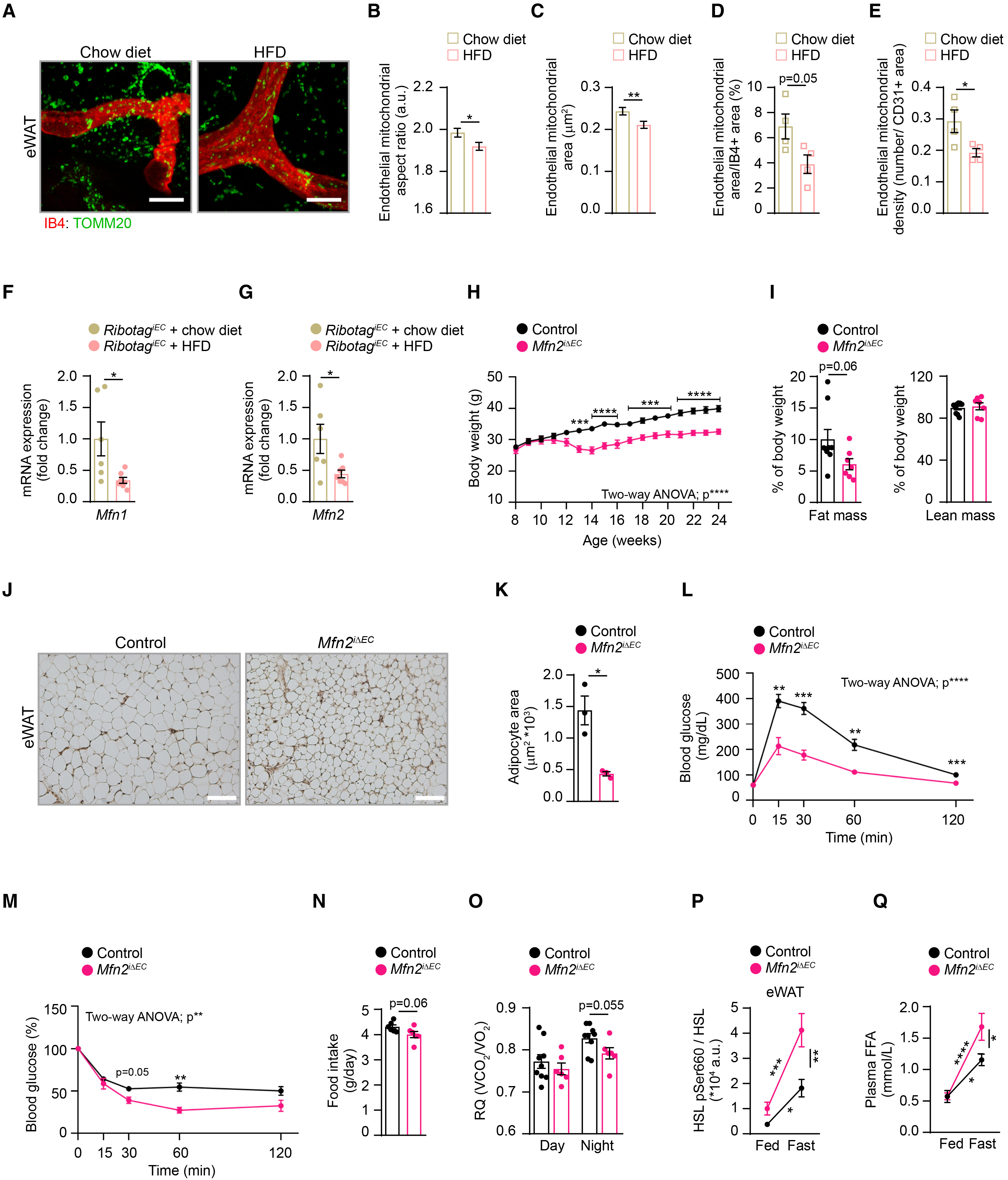 图1. ECs中Mfn2的缺失导致更苗条和代谢更健康的表型
图1. ECs中Mfn2的缺失导致更苗条和代谢更健康的表型
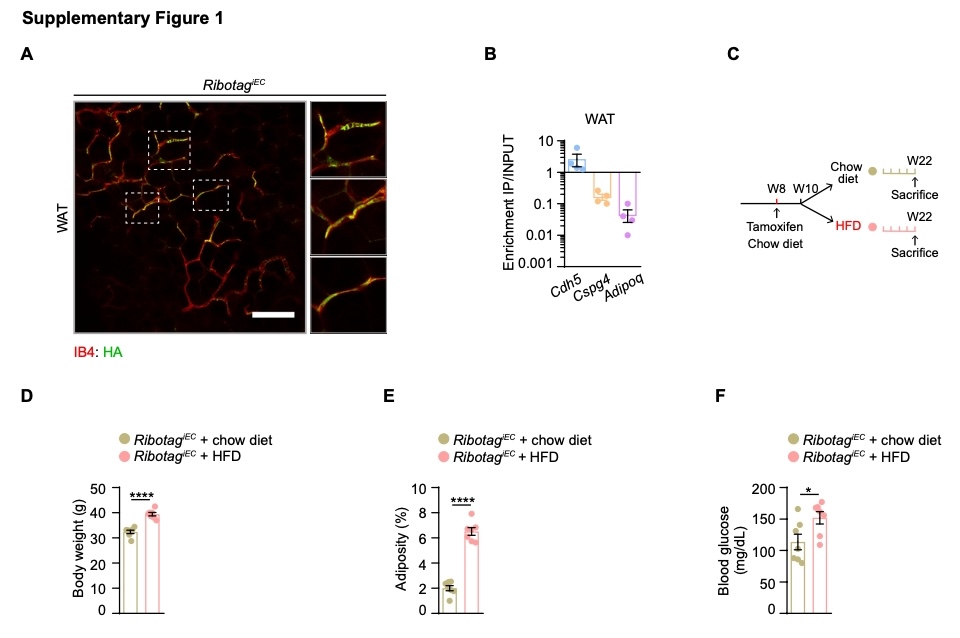 图S1. RibotagiEC小鼠模型的验证及高脂饮食下的代谢参数分析
图S1. RibotagiEC小鼠模型的验证及高脂饮食下的代谢参数分析
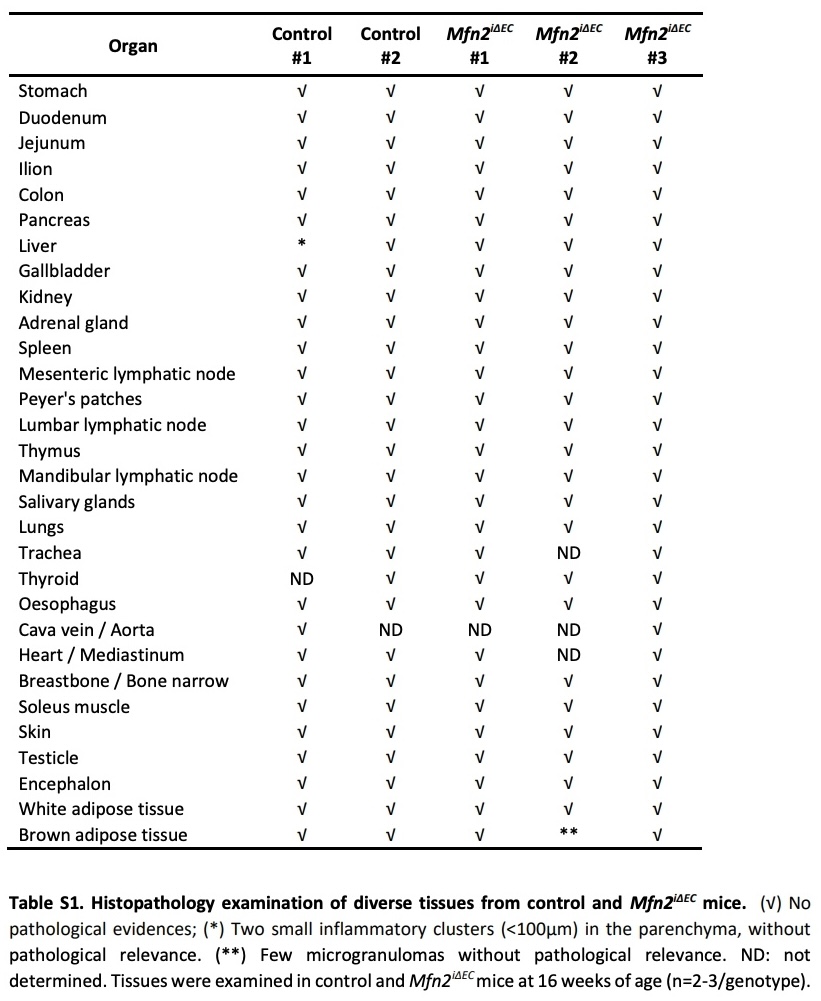 表1. 对照组和Mfn2iΔEC小鼠多个组织的组织病理学检查
表1. 对照组和Mfn2iΔEC小鼠多个组织的组织病理学检查
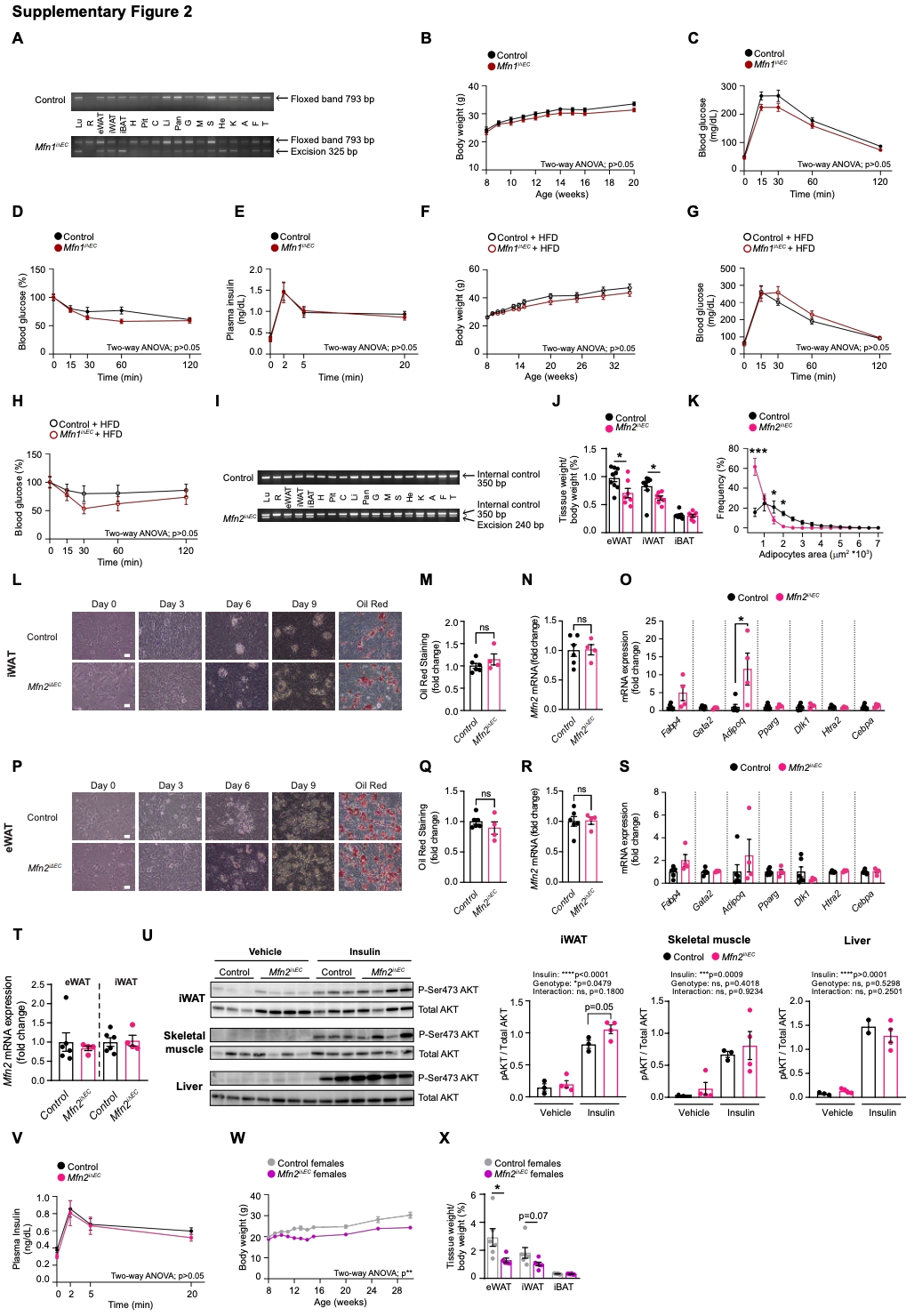 图S2. Mfn1iΔEC和Mfn2iΔEC小鼠的代谢特征分析
图S2. Mfn1iΔEC和Mfn2iΔEC小鼠的代谢特征分析
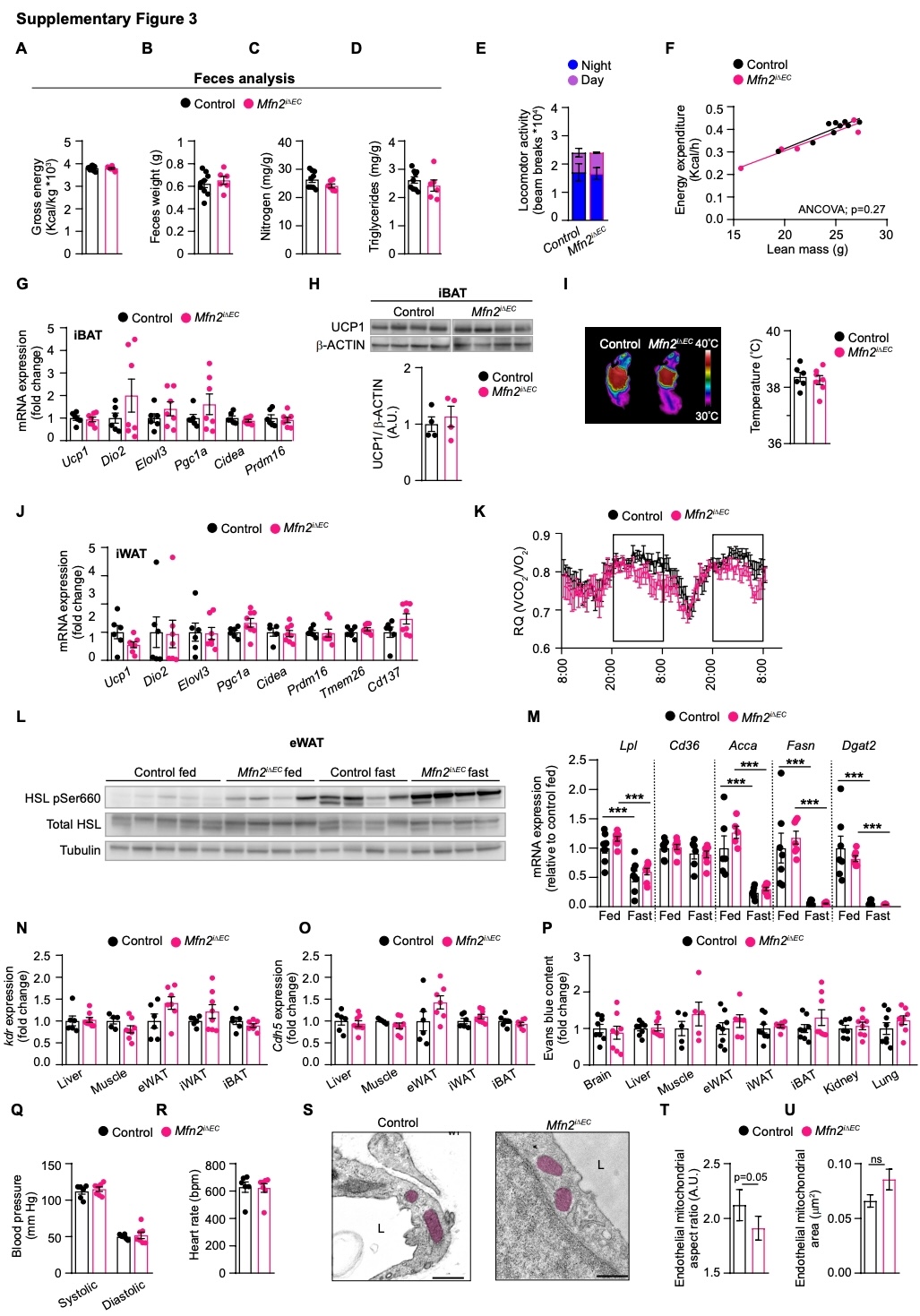 图S3. 内皮细胞中Mfn2的缺失与脂解作用增强相关
图S3. 内皮细胞中Mfn2的缺失与脂解作用增强相关
3、内皮细胞中Mfn2缺失导致血管网络保持不变,但蛋白翻译组特征发生显著改变
该实验室在过去的研究中证明,内皮细胞驱动的脂质氧化可促进血管生成。因此,研究人员探究了内皮细胞Mfn2
缺失是否会影响代谢组织中的血管分布。免疫荧光分析显示,Mfn2iΔEC小鼠与对照小鼠的各个组织的血管密度相似(图2A)(小编注:CD31/IB4并非代表比值,而是不同器官用的染料不同,肝脏和肌肉的冰冻切片用CD31染色,三种脂肪的整块组织块(whole-mount)用IB4染色(IB4是一种直接与荧光染料偶联的植物凝集素,通过特异性结合内皮细胞表面的α-D-半乳糖残基来标记内皮细胞,在图1A的脂肪染色图也是用的IB4),两者均为内皮细胞maker,用来反映血管密度,图中统计了血管占总面积的百分比。),且内皮标志物(血管内皮生长因子受体2(Kdr)和血管内皮钙粘蛋白5(Cdh5))的表达水平相似(图S3N和S3O)。血管的通透性(图S3P)、血压及心率均无差异(图S3Q和S3R)(小编注:Evansblue,伊文思蓝,是一种非膜渗透性的染料。当质膜受损,染料能进入细胞质和细胞核,从而将其染成蓝色,可用来检测细胞活力。此外,伊文思蓝在血液中与血浆白蛋白有很高的亲和力,所以伊文思蓝还能用来研究血脑屏障的完整性,由于正常状态下血浆白蛋白无法透过血脑屏障,所以染色后,具有完整血脑屏障的神经系统不着色。相反,如果神经系统血脑屏障被破坏,伊文思蓝就可以进入神经系统并使其着色。此处实验中,小鼠尾静脉注射伊文思蓝30分钟后,处死小鼠,取出各个代谢组织,置于500μL甲酰胺中55°C孵育24小时。经离心处理后,测定吸光度,然后通过计算转化为每毫克组织中结合的染料含量。研究人员在图S3P中检测了多个代谢器官中的血管通透性。结合血压和心率,共同证实内皮细胞Mfn2缺失不影响血管的结构和功能。)。接下来,研究人员探究了白色脂肪组织内皮细胞中线粒体的变化,电镜分析显示,Mfn2iΔEC小鼠的iWAT内皮细胞的线粒体长宽比降低,而线粒体面积没有变化(图S3S–S3U)(小编注:L为Lumen(血管腔),并且在线粒体(紫色标注)的另一侧,可以看到一层较深色的条索状结构基底膜。这种“腔-细胞-基底”的层级结构是内皮细胞的解剖特征。)。iWAT内皮细胞中与内质网接触的线粒体数量相似(对照组:76.3%±6.9%;Mfn2iΔEC组:73.0%±5.5%;p=0.71;n=22–24)。为了探究Mfn2缺失对分子特征的影响,研究人员构建了RibotagiEC(对照组)小鼠和RibotagMfn2iΔEC小鼠(小编注:如前所述,Ribotag是一种细胞类型特异性的翻译组分析工具,其核心原理是在目标细胞(如内皮细胞)的核糖体蛋白上引入HA标签,通过抗HA抗体免疫沉淀,直接从组织匀浆中特异性富集目标细胞中正在翻译的mRNA,而传统FACS/磁珠分选需要组织消化、分离细胞,过程容易引起应激转录改变。RiboTag则是先在体内给目标细胞核糖体打标签,再直接从组织匀浆里免疫沉淀,更接近原位状态。在这里研究者利用RiboTag构建了对照组(RibotagiEC)和Mfn2敲除组(RibotagMfn2iΔEC)小鼠,通过对比两组内皮细胞的翻译组,揭示了Mfn2缺失对内皮细胞翻译组的直接影响),并对WAT内皮细胞进行了组学分析(小编注:研究者利用RiboTag小鼠模型,无需将内皮细胞从组织中物理分离,而是通过以下步骤直接富集内皮细胞特异性翻译中mRNA用于测序:取白色脂肪组织匀浆后,加入小鼠单克隆抗HA抗体,该抗体特异性识别RiboTag小鼠内皮细胞核糖体上的HA标签,从而标记来源于内皮细胞的核糖体。加入磁珠,与抗HA抗体结合,形成“磁珠-抗体-核糖体-mRNA”复合物,经磁性分离和高盐缓冲液洗涤去除杂质后,提取总RNA进行测序。该方法避免了细胞分离过程中的应激干扰,能够真实反映内皮细胞在原位状态下的翻译活性)。qPCR结果证实了ECs中Mfn2的缺失。RNA-seq结果显示,共鉴定出1244个差异表达基因,其中大部分表达上调(图S4A)。与之前证明的Mfn2缺失可激活内质网未折叠蛋白反应(UPRER)的报道一致(参考文献:Ngoh, G.A. ∙ Papanicolaou, K.N. ∙ Walsh, K. Loss of mitofusin 2 promotes endoplasmic reticulum stress. J. Biol. Chem. 2012; 287:20321-20332),研究人员在RibotagMfn2iΔEC小鼠的WAT内皮细胞中也观察到UPRER相关基因表达增加(图S4B)。然而,给小鼠腹腔注射4-苯基丁酸(4-PBA,一种可恢复内质网蛋白质稳态、抑制内质网应激反应的化学保护剂),并未逆转代谢表型(图S4C–S4G),说明UPRER并非该表型的主要成因。
在1244个差异表达基因中,约7%的差异表达基因编码线粒体蛋白(图S4H),主要涉及代谢、线粒体中心法则和氧化磷酸化(OXPHOS)过程(图S4I)。基因功能富集分析也证实了OXPHOS和脂肪酸代谢相关基因的富集(图2B和S4J)。这与Mfn2iΔEC小鼠倾向于使用脂质作为燃料(图1O–1Q、S3K和S3L)的特征一致,提示内皮细胞Mfn2的缺失会促进脂质氧化。并且,活性氧(ROS)通路也是显著富集的类别之一(图2B)。转录因子结合位点分析发现,过氧化物酶体增殖物激活受体α(PPARα)、PPARγ和核因子E2相关因子2(NRF2;由Nfe2l2编码)的结合位点存在富集(小编注:首先将差异表达基因(DEGs)输入Enrichr在线分析工具,选择“TRRUST Transcription Factors 2019”数据库进行转录因子富集分析。TRRUST是一个专门收录转录因子与其靶基因调控关系的数据库,通过该分析可获得每个转录因子靶基因集在DEGs中的富集显著性(p值)和综合得分。根据p值对所有转录因子进行排序,选取排名前十的转录因子,将p值转换为-log10(p-value)后进行绘图)(图2C),这些转录因子在脂质代谢、线粒体功能和抗氧化反应中发挥关键作用。Mfn2iΔEC小鼠内皮细胞中线粒体蛋白质稳态(UPRmt)的标志基因表达也升高(图S4K)。然而,白色脂肪组织内皮细胞中发生变化的标志基因在脂肪细胞中则没有发生变化(图S4L),这表明MFN2基因缺失后,发生了内皮细胞特异性的转录调控。Mfn2iΔEC小鼠的骨骼肌内皮细胞(而非肝脏内皮细胞)表现出与WAT内皮细胞相似的基因表达变化(小编注:本小节评估内皮Mfn2缺失对代谢组织的影响,研究了肝脏、骨骼肌、eWAT、iWAT和iBAT的血管结构、功能以及内皮细胞分子特征。脂肪组织内皮细胞的发生基因表达变化且脂肪细胞本身未受影响,说明敲除后发生的是内皮细胞特异性的适应反应。骨骼肌内皮细胞中发生相似的变化肝脏则未发生,也体现了内皮细胞的组织特异性变化,但作者没有进一步研究发生这一现象的原因。我们猜测这是由内皮细胞的组织特异性导致的,如脂肪与骨骼肌对脂质代谢和线粒体氧化磷酸化的依赖程度更高。此外,肝脏作为代谢器官,其代谢稳态的改变可能更多地依赖于肝细胞的功能,而非肝内皮细胞。或是肝脏内皮细胞中可能存在其他融合蛋白(如Mfn1或OPA1)的高表达,在Mfn2缺失时能够完全代偿其功能,而在脂肪或肌肉内皮中, Mfn2 的功能可能是不可替代的)(图S4M和S4N)。
综上所述,内皮细胞Mfn2缺失虽不影响血管结构或功能,但可重塑白色脂肪组织内皮细胞的翻译组。尽管这些特征中没有任何一个能单独解释Mfn2iΔEC小鼠的代谢表型改善,但综合来看,它们与线粒体低毒兴奋效应反应的特征相一致。
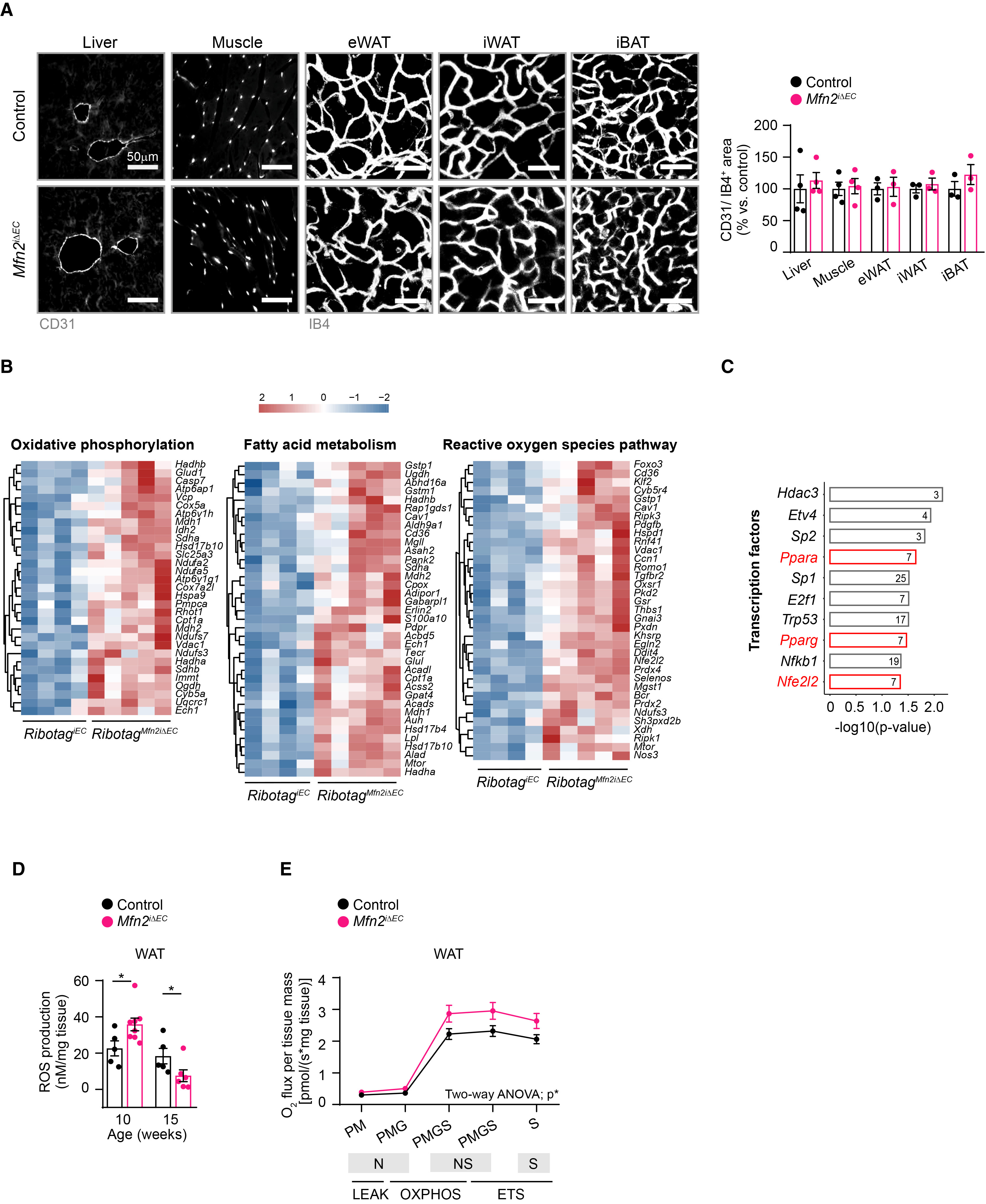
图2. 内皮Mfn2缺失导致血管线粒体发生适应性变化,引发线粒体低毒兴奋效应
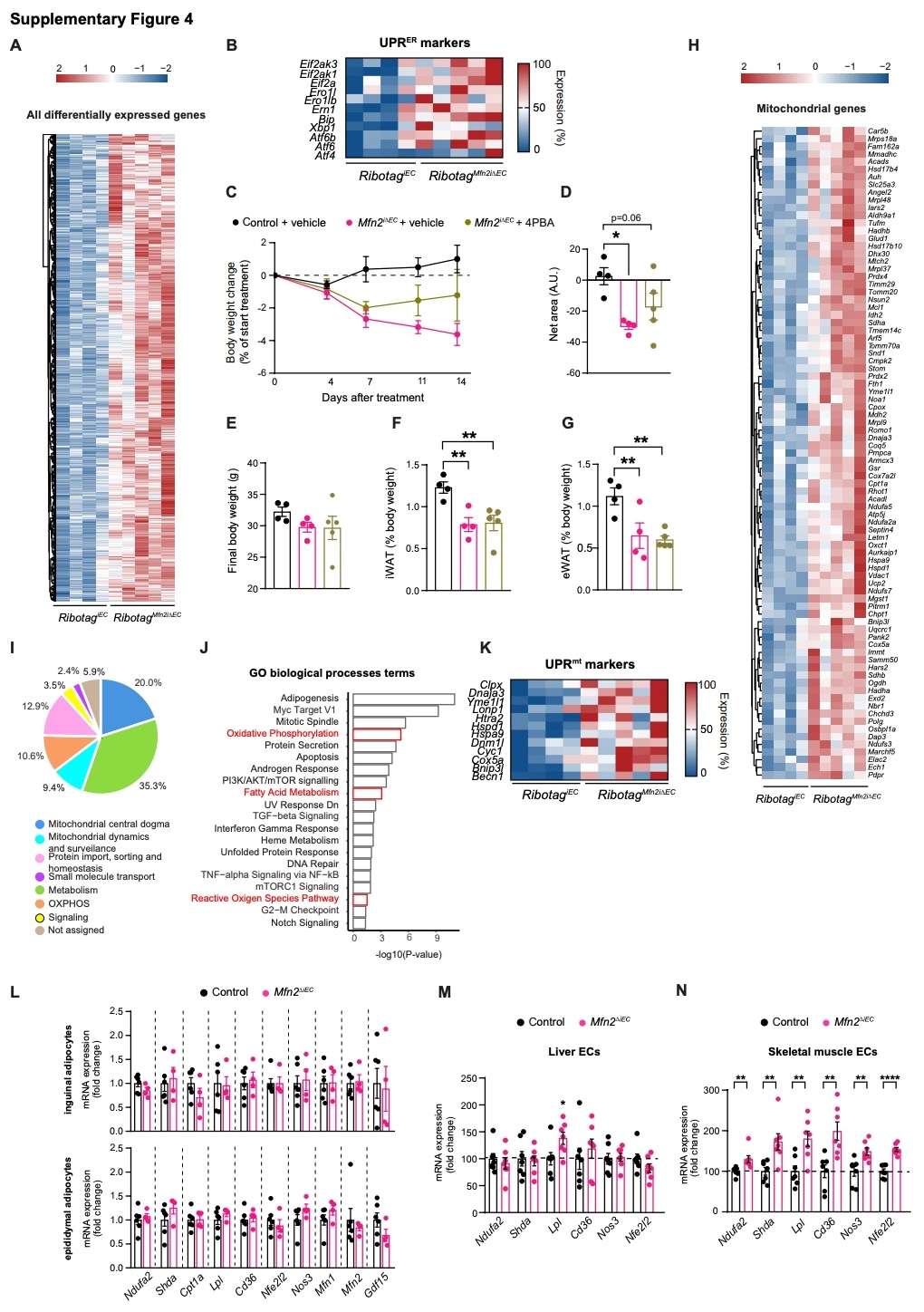 图S4. 内皮细胞中Mfn2的缺失会改变内皮细胞的翻译组
图S4. 内皮细胞中Mfn2的缺失会改变内皮细胞的翻译组
4、Mfn2的缺失会导致血管线粒体发生适应性改变,从而引发线粒体低毒兴奋效应
线粒体低毒兴奋效应(Mitohormesis)是细胞应对代谢应激时产生的一种适应性反应,它会激活保护性机制,最终增强细胞功能、代谢健康和机体抵抗力。线粒体低毒兴奋效应的一个特征,是线粒体活性氧的瞬时增加,这会诱导防御机制的启动来降低活性氧水平。为了确定内皮细胞Mfn2缺失是否会触发线粒体低毒兴奋效应,研究人员首先在10周龄和15周龄小鼠的离体白色脂肪组织中检测了活性氧水平。结果显示,与对照组相比,Mfn2iΔEC小鼠WAT的活性氧水平呈现先升高后降低的趋势(图2D),而在肝脏或骨骼肌中未观察到这一现象(图S5A和S5B)。
线粒体低毒兴奋效应的另一个特征是在活性氧生成减少的同时,增强线粒体活性,高分辨率呼吸测定显示,与对照小鼠相比,Mfn2iΔEC小鼠WAT的复合物I及复合物II介导的呼吸链活性更强(图2E)(小编注:N代表NADH氧化呼吸链,S代表琥珀酸氧化呼吸链。在呼吸链中,复合体Ⅰ(NADH:辅酶Q氧化还原酶复合体)从NADH得到两个电子传递给辅酶Q。复合体Ⅱ(琥珀酸脱氢酶)从琥珀酸得到电子传递给辅酶Q。辅酶Q将从这两个复合体接受的电子传递给复合体Ⅲ(辅酶Q:细胞色素C氧化还原酶复合体),最后传递给氧。2E实验叫做高分辨率线粒体呼吸测定法,使用OroborosOxygraph-2k仪器测定,采用新鲜分离的组织,将组织放入Oxygraph中,平衡后添加底物,检测氧通量。包括LEAK呼吸(线粒体不进行ADP磷酸化时,质子漏所驱动的耗氧量)测定:添加苹果酸和丙酮酸(复合体I连接的底物),无ADP存在;OXPHOS(氧化磷酸化)状态测定:添加ADP+MgCl2和细胞色素C,随后添加谷氨酸(复合体Ⅰ链接底物)和琥珀酸(复合体Ⅱ链接底物)进行测定。并通过滴定FCCP(氧化磷酸化解偶联剂)来评估电子传递系统的最大呼吸能力:逐渐增加培养基中FCCP的浓度来滴定FCCP,每次添加后,呼吸速率应保持至少1分钟以达到平衡,当后续添加FCCP不会进一步增加呼吸时,该值通常被认为是最大呼吸能力。使用鱼藤酮抑制复合体I,测定由复合体Ⅱ途径驱动的电子传递系统的最大呼吸能力。使用抗霉素A抑制复合体III,由于复合物III是来自复合物I或复合物II的电子所需的受体,因此加入抗霉素A后阻止了线粒体电子传递,测得的耗氧率被视为背景耗氧量,而非线粒体呼吸,被从先前的值中减去用于分析。氧气通量值表示为每秒钟每毫克组织湿重的耗氧量(皮摩尔氧气/秒·毫克组织)。参考文献:C. Cantó, P.M. Garcia-Roves. High-Resolution Respirometry for Mitochondrial Characterization of Ex Vivo Mouse Tissues. Curr. Protoc. Mouse Biol., 5 (2015), pp. 135-153, 10.1002/9780470942390.mo140061.
基于Oroboros Oxygraph-2k仪器的高分辨率呼吸测定法利用经典的电极技术检测氧气浓度变化,其准确度更高、通量更低、样品量需求较大,适合线粒体的深入研究,如底物、抑制剂、解偶联剂的复杂滴定;组织活检(如肌肉、肝脏、脂肪)或分离的线粒体,能够通过精确的耗氧率来评估线粒体功能。Seahorse细胞能量代谢分析仪通量高、样品量需求极少。可微孔板一次平行检测大量样品,主要获得基础呼吸、ATP相关呼吸、最大呼吸、非线粒体呼吸等标准参数,能同步评估线粒体呼吸和糖酵解,但背景值波动相对较大)。相比之下,其他组织中未检测这一现象(图S5C和S5D),说明WAT对代谢应激的适应存在组织特异性。综上所述,内皮细胞Mfn2的缺失会促进细胞在分子和功能层面的调整,这与WAT中线粒体低毒兴奋效应的激活相一致。尽管骨骼肌内皮细胞的翻译组也表现出一定的变化(图S4N),但这些变化并未导致活性氧生成或线粒体呼吸的改变(图S5B–S5D)。
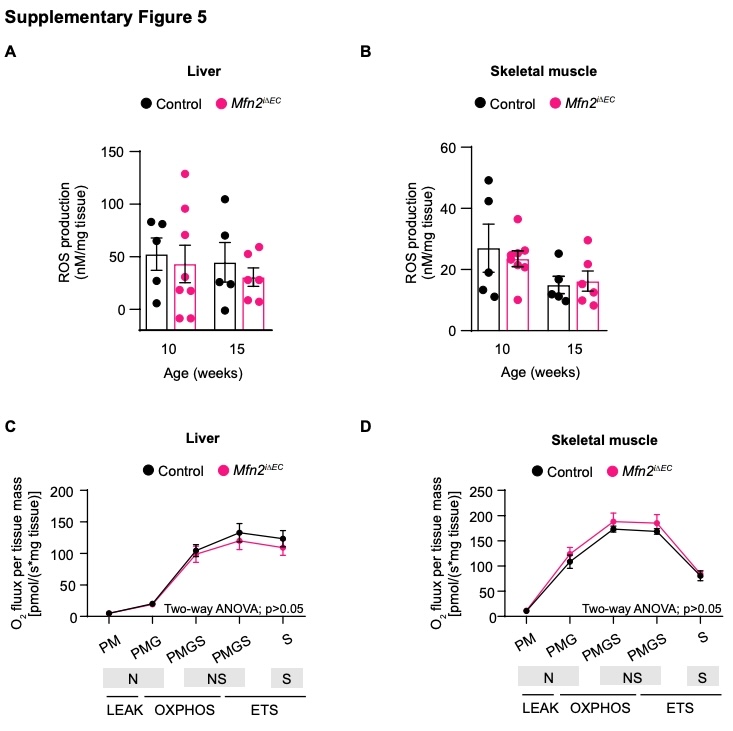
图S5. 内皮细胞缺失Mfn2不会改变肝脏或骨骼肌中活性氧的产生和线粒体呼吸
5、GDF15由ECs产生和分泌
鉴于在Mfn2iΔEC小鼠中观察到的表型,研究人员推测内皮细胞相关的线粒体低毒兴奋效应可能由某种内皮细胞来源的分泌蛋白所驱动。为了筛选可能的驱动因子,研究人员对表达E4ORF1的人脐静脉内皮细胞(HUVECs)的分泌蛋白谱进行了分析,E4ORF1能使细胞在无血清条件下长期存活和生长(小编注:HUVEC(人脐静脉内皮细胞)是从新生儿脐带静脉中分离出来的原代内皮细胞,是最经典的模型细胞之一,但原代HUVEC在缺乏血清和细胞因子的培养环境中会迅速凋亡,失去内皮细胞特性。在原代细胞基础上,转染腺病毒E4ORF1基因并筛选获得E4ORF1稳定表达细胞株后,HUVEC在能够无血清/无细胞因子条件下的维持细胞形态,保留内皮细胞功能。参考文献:M. Seandel, J.M. Butler, H. Kobayashi, A.T. Hooper, I.A. White, F. Zhang, E.L. Vertes, M. Kobayashi, Y. Zhang, S.V. Shmelkov, et al. Generation of a functional and durable vascular niche by the adenoviral E4ORF1 gene. Proc. Natl. Acad. Sci. USA, 105 (2008), 19288-19293, 10.1073/pnas.0805980105.)。研究人员收集了E4ORF1阳性的HUVECs培养基(CM),并进行蛋白质组学分析(图3A),共鉴定出818种蛋白质,且CM组与非条件培养基对照组样品间呈现明显的聚类差异(图3B和3C)。值得注意的是,有227种蛋白在CM中显著上调(图3C)。在这227种上调的蛋白质中,有116种仅在CM组中被检测到(图3C,高亮矩形区域),表明它们是由内皮细胞特异性分泌的。
运用SignalP5和Secretome 2.0软件进行预测后,研究人员鉴定出113种经典的内皮细胞分泌蛋白(图3D)。剩余的114种蛋白质与外泌体(ExoCarta)和细胞外囊泡(Vesiclepedia)数据库进行比对,发现分别有105种和110种蛋白与二者存在重叠(图3D)。基因集富集分析将这些蛋白归类到与细胞外空间、膜结构、囊泡及膜包围腔室相关的类别,进一步证实了它们的分泌蛋白属性(图3E)。接下来,研究人员利用CellPhoneDB来分析细胞间相互作用。从最初鉴定出的227种蛋白质中,研究人员筛选出其中29种蛋白,它们被鉴定为配体、细胞外基质蛋白或受体(图3F)。其中,16种是目前已知的配体(图3G),其中许多蛋白在血管生物学中发挥着明确作用。在这些配体中,生长分化因子15(GDF15)引起了研究人员的关注(图3G),在之前的研究中发现,GDF15是一种能够响应代谢应激和介导线粒体低毒兴奋效应的关键细胞因子,并且有益于机体的代谢。首先,研究人员验证了内皮细胞在体外培养条件下能够产生并分泌GDF15(图3H)。其次,为了探究在体内GDF15是否也由内皮细胞产生,研究人员将Gdf15fl/fl小鼠与Pdgfb-iCreERT2小鼠杂交,构建了内皮细胞特异性Gdf15敲除小鼠(Gdf15iΔEC小鼠),使多种组织中的内皮细胞Gdf15表达降低约80%(图S6A和S6B),但不改变基础血浆GDF15水平(图3I)。鉴于GDF15在脂多糖等应激条件下会迅速升高,研究人员给予小鼠LPS刺激,发现与对照组相比,Gdf15iΔEC小鼠的循环GDF15水平显著降低(图3J)。这些结果表明,内皮细胞在应激状态下会提高全身的GDF15水平。
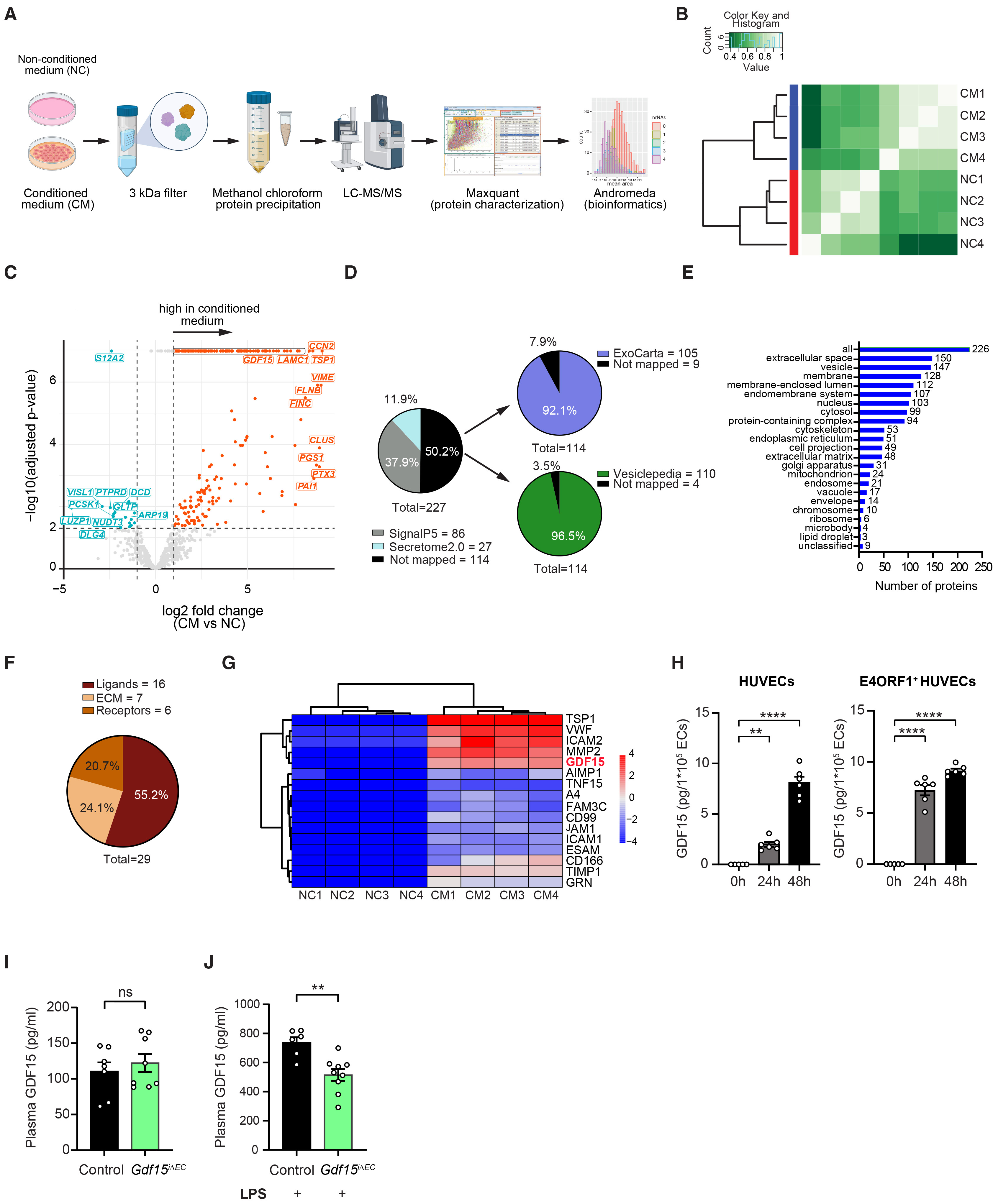
图3. 内皮细胞合成和分泌GDF15
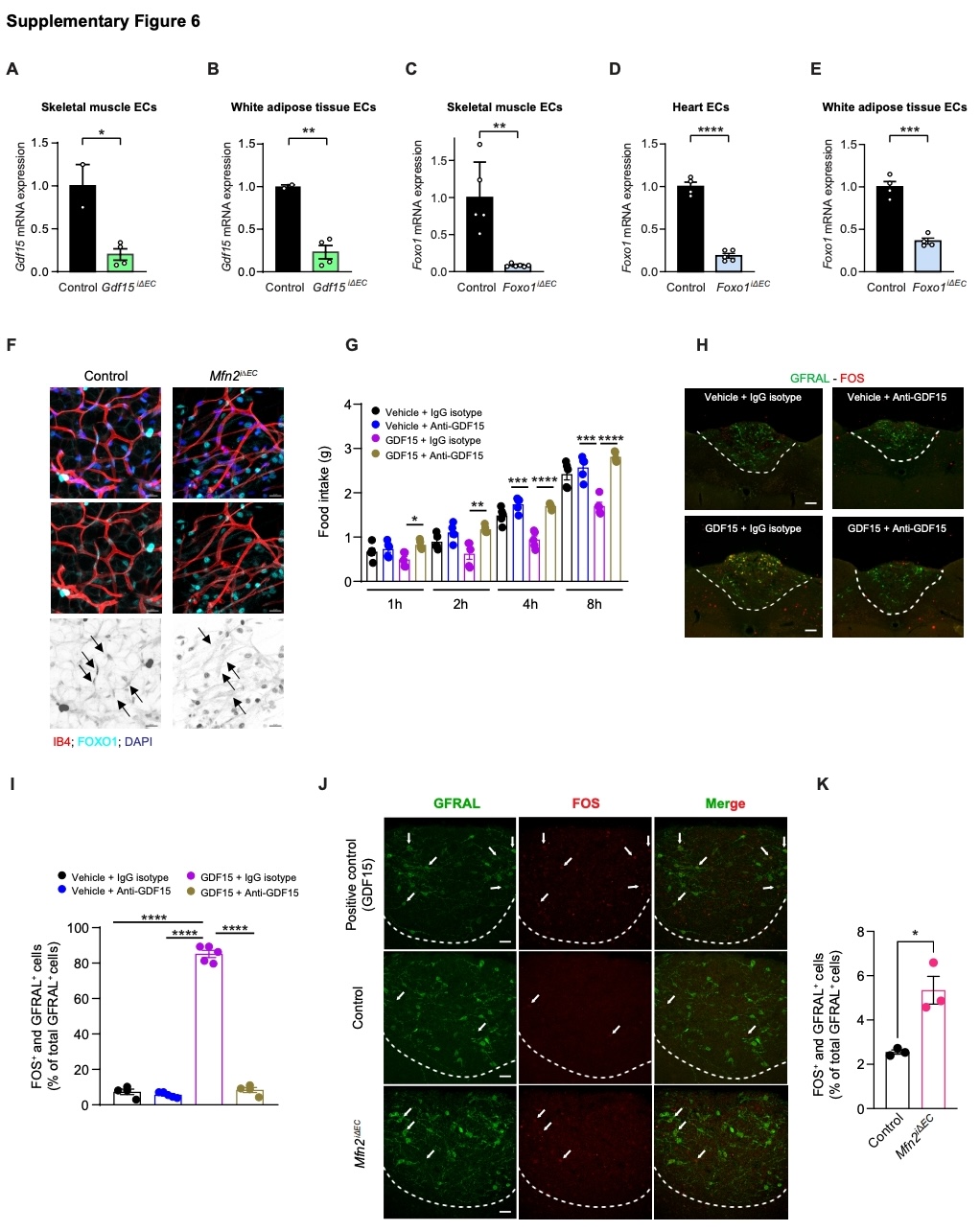 图S6. Gdf15i∆EC和Foxo1i∆EC小鼠模型的验证,以及GDF15中和抗体策略疗效的评估
图S6. Gdf15i∆EC和Foxo1i∆EC小鼠模型的验证,以及GDF15中和抗体策略疗效的评估
6、内皮细胞以FOXO1依赖的方式分泌GDF15
接下来,研究人员研究了GDF15水平的调控机制。鉴于内皮细胞在正常状态下对循环GDF15水平并无贡献,研究人员推测在稳态条件下GDF15的表达可能会受到抑制。在稳态下,内皮细胞大部分处于静息状态,并且这是由NOTCH1和FOXO1通路调控的。因此,研究人员探究了这些通路是否参与GDF15的产生。结果显示,在HUVECs中用NOTCH1的配体DLL4激活NOTCH信号通路,或用γ-分泌酶抑制剂DAPT来抑制NOTCH信号通路,均未影响GDF15的mRNA水平(图4A和4B)。相比之下,用四环素诱导表达系统激活FOXO1可降低HUVECs和E4ORF1+ HUVECs中的GDF15 mRNA水平及其分泌(图4C–4F)。下游靶基因表达的增加证实了FOXO1的激活(图4C–4E)。相反,使用AS1842856抑制FOXO1则导致内皮细胞中GDF15表达升高(图4G)以及培养基中GDF15的积累(图4H)。值得注意的是,研究人员使用丝裂霉素C(抑制细胞增殖)处理HUVECs后,仍出现了相同的结果(图4I和4J),表明FOXO1介导的这些变化与内皮细胞的增殖无关。为了探究FOXO1在体内调控GDF15中的作用,研究人员构建了内皮细胞特异性Foxo1敲除的小鼠模型(Foxo1iΔEC小鼠)。基因表达分析显示,从多种组织分离的内皮细胞中Foxo1 mRNA均下调(图S6C–S6E)。值得注意的是,内皮细胞Foxo1缺失导致多种组织的内皮细胞中的Gdf15表达显著增加(图4K–4M)。血清和尿液的GDF15水平进一步证实,内皮细胞中Foxo1的缺失可增加循环GDF15水平(图4N和4O);尿液中GDF15水平的升高,表明肾小球滤过负荷增加以及近端肾小管重吸收部分饱和,而非肾功能障碍所致。与此一致的是,在体内注射AS1842856药物抑制FOXO1同样可提高循环GDF15水平(图4P)。综上所述,内皮细胞以FOXO1依赖的方式产生并分泌GDF15。
拓展阅读
内皮细胞的静息态和激活态
血管内皮细胞(Endothelial Cells, ECs)是位于血管内壁的单层细胞,是血液与组织之间的关键屏障。在生理稳态下,绝大多数血管内皮细胞处于一种被称为“静息”(Quiescence)的特殊状态,其特征是增殖停滞、代谢水平低、屏障功能稳定。然而,在组织损伤、炎症或肿瘤生长等病理条件下,内皮细胞可被迅速“激活”(Activation),表现为增殖、迁移、通透性增加和促炎表型。这种从静息到激活的精确转变是维持血管稳态和应对外界刺激的基础。
NOTCH1通路:维持内皮细胞静息的“刹车”系统
Notch信号通路是一个在进化上高度保守的细胞间通讯系统,对细胞命运决定、增殖和分化至关重要。在血管系统中,NOTCH1信号被证实是维持内皮细胞静息、确保血管稳定性的关键“刹车”分子。当内皮细胞表面的NOTCH1受体与其相邻细胞表面的配体(如DLL4或Jagged1)结合后,受体发生构象变化,其胞内结构域(NICD)被蛋白酶切割并释放。随后,NICD进入细胞核,与转录因子RBP-Jκ结合,形成转录激活复合体,启动下游靶基因(如HES和HEY家族基因)的表达。这些靶基因大多是转录抑制因子,它们通过多种机制来抑制细胞增殖,从而促进细胞静息。当该通路功能缺失或受到抑制时,内皮细胞会自发地脱离静息状态,进入细胞周期,导致血管过度增殖和结构不稳定。
FOXO1通路:守护内皮细胞静息的“看门人”
叉头转录因子家族成员FOXO1是另一个调控内皮细胞静息的核心因子,被誉为静息状态的“看门人”(Gatekeeper)。与NOTCH1主要通过调控细胞周期来维持静息不同,FOXO1更多地从代谢和基因水平上对细胞状态进行调控。FOXO1的活性受到上游PI3K-AKT信号通路负反馈调控。在静息状态下,由于缺乏生长因子刺激,AKT激酶活性较低,使得具有活性的FOXO1能够停留在细胞核内,发挥其转录因子的功能。当细胞受到VEGF等生长因子刺激时,PI3K-AKT通路被激活,AKT会磷酸化FOXO1,导致其被输出到细胞质中并失活,从而解除了对细胞增殖和激活的抑制。
内皮细胞的激活态(Activated State)
当机体需要生成新血管(如伤口愈合、胚胎发育)或应对炎症、缺氧等刺激时,静息的内皮细胞可以被迅速激活。激活态是一种高能耗、高动态的功能状态,其特征与静息态截然相反,主要表现在以下几个方面:
细胞增殖与迁移:激活的内皮细胞重新进入细胞周期,开始快速分裂和定向迁移,形成新的血管,这一过程被称为血管生成(Angiogenesis)。
高代谢活性:为了支持快速的增殖和迁移,细胞代谢(主要指糖酵解途径)被显著上调,为细胞的生物合成和能量需求提供原料与动力。
促炎与促血栓表型:细胞表面开始大量表达粘附分子(如ICAM-1, VCAM-1),招募白细胞至症部位。同时,细胞因子和趋化因子的分泌增加,呈现促炎状态。
通透性增加:细胞间的连接被重塑和削弱,导致血管屏障通透性增加,允许血浆蛋白和白细胞更容易地渗出到周围组织。
血管内皮生长因子(VEGF)、成纤维细胞生长因子(FGF)等促血管生成因子以及肿瘤坏死因子-α(TNF-α)等炎症因子是诱导内皮细胞激活的关键化学信号。虽然内皮细胞激活在生理过程中不可或缺,但其失控的、慢性的激活是许多重大疾病,如动脉粥样硬化、糖尿病血管病变和癌症进展的核心病理环节。
内皮细胞从静息到激活的精确调控是血管生物学的核心问题。FOXO1和NOTCH这两个通路共同形成了一个稳健、多层次的调控网络。这个网络确保了在生理稳态下内皮细胞能够可靠地维持静息,同时又保留了在需要时迅速响应并激活的
参考文献:
[1] Wilhelm K, Happel K, Eelen G, et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature. 2016;529(7585):216-220.
[2] Andrade J, Shi C, Costa ASH, et al. Control of endothelial quiescence by FOXO-regulated metabolites. Nat Cell Biol. 2021;23(4):413-423.
[3] Kalucka J, Bierhansl L, Conchinha NV, et al. Quiescent Endothelial Cells Upregulate Fatty Acid β-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab. 2018;28(6):881-894.e13.
[4] Augustin HG, Koh GY. A systems view of the vascular endothelium in health and disease. Cell. 2024;187(18):4833-4858.
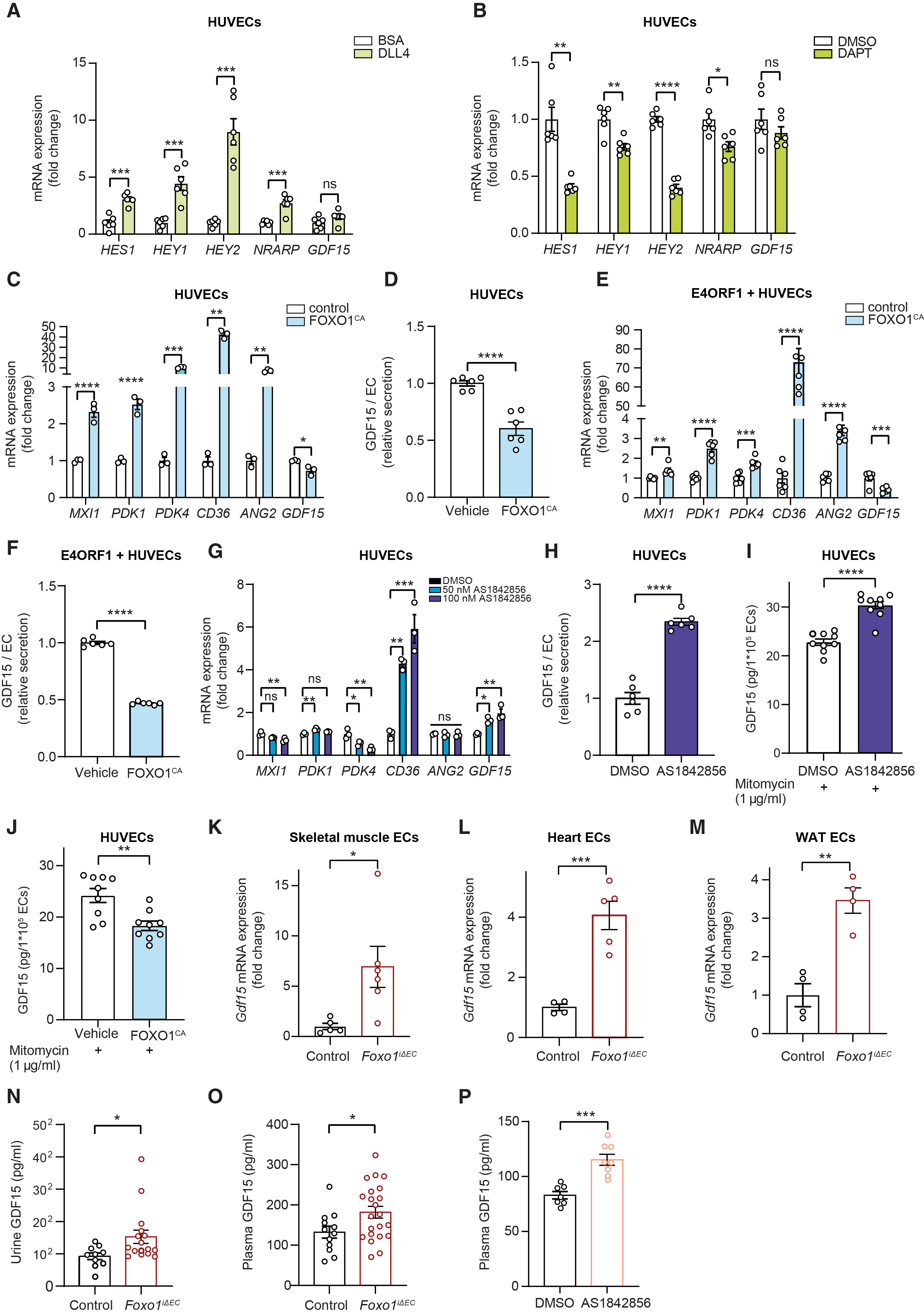 图4. FOXO1负向调控ECs中GDF15的表达
图4. FOXO1负向调控ECs中GDF15的表达
7、GDF15有助于维持Mfn2iΔEC小鼠的代谢健康
接下来,研究人员探究了GDF15表达升高是否介导了内皮细胞Mfn2缺失小鼠的表型。研究人员发现在HUVECs细胞中敲低MFN2后,GDF15的表达水平显著升高(图5A和5B)。与小鼠模型相似,这些细胞也表现出NFE2L2和活性氧水平的增加(图5C和5D)。重要的是,HUVECs中MFN2的缺失与FOXO1的抑制相关,表现为FOXO1 Ser256位点磷酸化(一种失活修饰)水平升高(图5E)。此外,研究人员分析了Mfn2iΔEC小鼠分离的WAT内皮细胞中,FOXO1在细胞核(活性)和细胞质(非活性)中的分布(图5F和S6F),发现FOXO1在细胞质中的积累增加,这说明FOXO1的失活可能解除了FOXO1对GDF15的转录抑制作用,从而解释了GDF15的上调。
为了探究在Mfn2缺失背景下GDF15在体内的功能,研究人员首先利用已有的RNA-seq数据集进行分析,发现在对照组小鼠中几乎检测不到Gdf15,而Mfn2iΔEC小鼠的WAT内皮细胞中Gdf15呈升高趋势(对照组:0.47 ± 0.47 CPM vs. Mfn2 iΔEC :11.86 ± 5.61 CPM;p = 0.12)。研究人员利用另一批独立的RibotagiEC和RiboTagMfn2iΔEC小鼠,证实了敲除MFN2的WAT内皮细胞中Gdf15表达升高(图5G),但肝脏内皮细胞或肝脏实质组分中未发现这一变化,肝实质组分的Gdf15 mRNA水平高于内皮细胞,这与肝实质细胞是肝脏Gdf15主要来源的结论一致(图5H)。Gdf15 mRNA的上调伴随着循环GDF15水平的短暂升高,在11-13周龄达到峰值(图5I)。血浆成纤维细胞生长因子21(FGF21,另一种参与多种代谢功能的线粒体应激激素)水平则无变化(图5J)。
接下来,研究人员通过在Mfn2iΔEC小鼠体重下降初期阶段通过中和抗体来阻断GDF15信号通路,来探究GDF15在该小鼠表型中的作用(图5K)。抗体的有效性通过结合亲和力研究、细胞生物活性测定(文章中未展示)以及在野生型小鼠的体内实验进行了验证。在野生型小鼠的体内实验中,研究人员发现该抗体可完全逆转GDF15的抑制食欲作用并阻断GDNF家族受体α样(GFRAL)信号通路(图S6G–S6I)。使用抗GDF15抗体处理后,Mfn2iΔEC小鼠的体重下降幅度减缓,脂肪量得到部分恢复(图5L–5N)。
GDF15通过后脑最后区(AP,Area postrema)和孤束核(NTS,nucleus of the solitary tract)的GFRAL受体促进体重减轻,GFRAL和FOS(FOS阳性代表神经元被激活)的免疫定位结果显示,Mfn2iΔEC小鼠中激活的GFRAL+神经元比例更高(图5O和5P),这与血浆GDF15水平升高相一致(图5I)。在20周成年小鼠中,尽管对照小鼠和Mfn2iΔEC小鼠的循环GDF15水平相同,Mfn2iΔEC小鼠中也同样观察到GFRAL信号通路的增强(图S6J和S6K)。
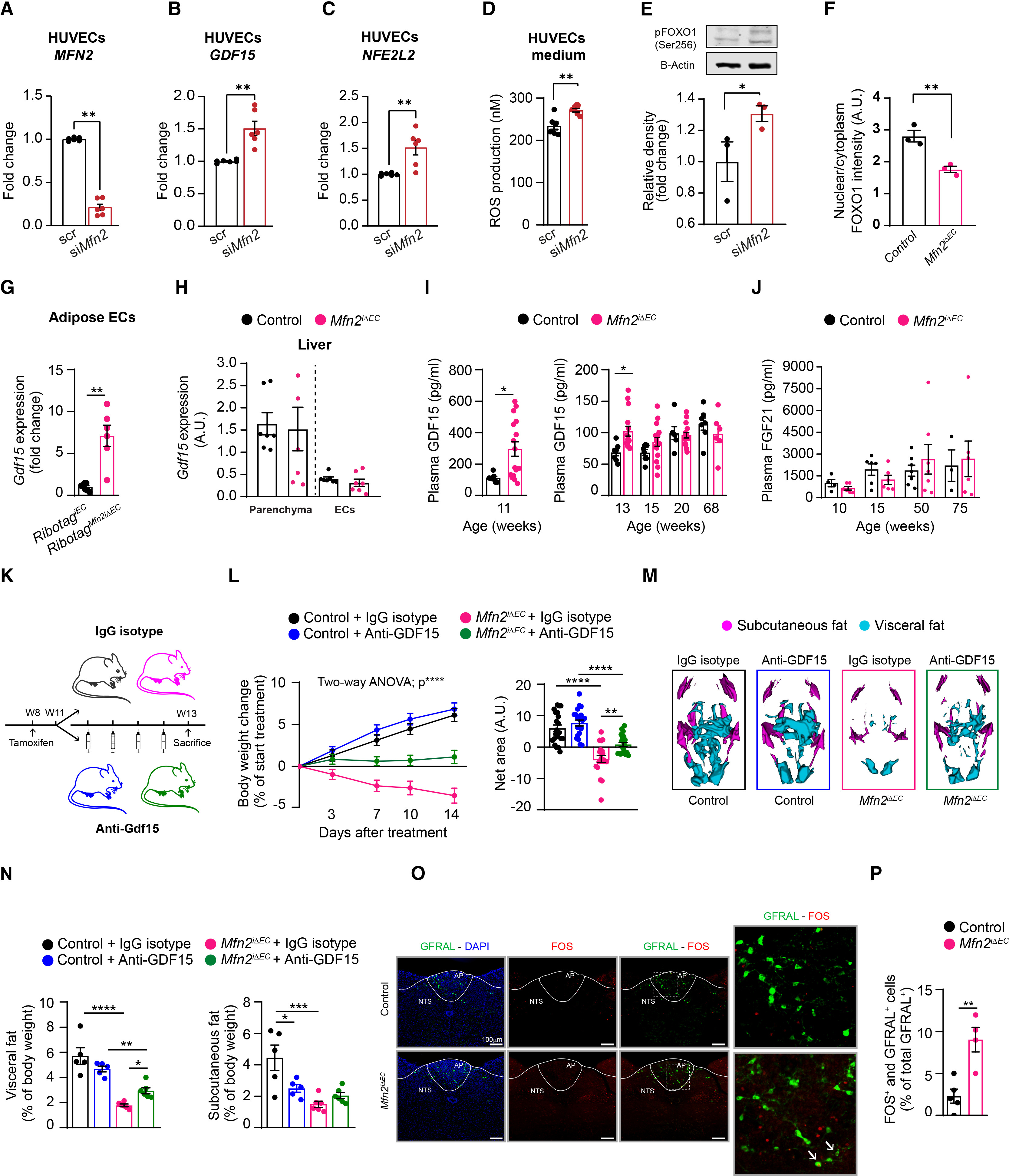 图5. 内皮细胞Mfn2的缺失增加了GDF15的表达,并部分介导了瘦型表型
图5. 内皮细胞Mfn2的缺失增加了GDF15的表达,并部分介导了瘦型表型
8、内皮细胞Mfn2的缺失可增强对饮食诱导性肥胖的抵抗力,并改善衰老表型
既往研究证明,线粒体低毒兴奋效应可改善多种模式生物(包括小鼠)的代谢,并促进健康衰老。为此,研究人员首先探究了线粒体低毒兴奋效应是否能够改善Mfn2iΔEC小鼠饮食诱导的肥胖。结果显示,内皮细胞Mfn2缺失使小鼠对饮食诱导的肥胖产生抵抗力,并改善了葡萄糖代谢(图6A–6G和S7A–S7D)。此外,在已经肥胖的情况下,通过他莫昔芬诱导内皮细胞Mfn2缺失仍可使体重、脂肪量和葡萄糖代谢恢复正常(图6H–6O和S7E–S7H)。这些结果表明,血管内皮细胞Mfn2缺失既可预防、也可逆转饮食诱导的肥胖。
接下来,研究人员探究了内皮细胞Mfn2缺失对衰老相关的影响。结果显示,内皮细胞Mfn2缺失并未影响突变小鼠的平均寿命或最大寿命(图S7I)。随后,研究人员通过检测衰老相关生物指标,分析了年轻(16-20周)和衰老(70-75周)的对照和Mfn2iΔEC小鼠中的衰老指标。结果显示,相比于WT小鼠,Mfn2iΔEC小鼠中与衰老相关的体重下降被推迟(图7A、7B和S7J)。两组衰老小鼠的葡萄糖代谢参数无差异,这可能是由于两组衰老小鼠的体重相当(图S7K和S7L)。虽然在年轻和衰老小鼠中,WT和Mfn2iΔEC小鼠的股骨结构都相似(图S7M–S7P),但在衰老的Mfn2iΔEC小鼠中,血液学参数没有出现明显的衰老相关衰退(图7C–7E)。此外,Mfn2iΔEC小鼠尿液中与衰老相关的白蛋白与肌酐的比率(ACR,肾脏损伤的标志物)降低(图7F)。
大脑功能会随年龄增长而逐渐衰退,影响感觉感知、认知表现和运动协调。研究人员对神经学参数的标准定性分析显示,衰老WT小鼠出现了轻度的年龄相关功能衰退,而衰老Mfn2iΔEC小鼠的神经学参数与年轻时相近(图S7Q)。此外,衰老WT小鼠的自主活动、协调性和平衡能力(图7G–7K、S7R和S7S)以及认知能力(图7L)下降,而Mfn2iΔEC小鼠未出现明显下降(小编注:Rotarod(转棒仪)和Balance Beam(平衡木)是两种常用的小鼠运动协调与平衡能力测试方法,虽然都用于评估神经运动功能,但其测试原理、测量指标、敏感性以及所反映的神经机制有明显区别。1、测试原理:Rotarod是持续运动状态下维持平衡,而Balance Beam是静态与动态结合的运动协调。2、主要测量指标:Rotarod是测量小鼠在加速或匀速转棒上的停留时间(潜伏期),而Balance Beam是测量小鼠穿过横梁的时间、步态、滑足次数。3、Rotarod是持续性运动适应实验,而Balance Beam是精细运动控制与平衡实验。4、两种实验检测的神经功能不同,Rotarod侧重整体运动协调、小脑功能、运动学习;而Balance Beam侧重皮层运动整合、精细运动、后肢功能。5、Rotarod对轻度运动障碍、运动学习能力敏感,而Balance Beam对精细运动缺陷、感觉运动整合障碍敏感。这两类实验常联合使用以全面评估小鼠运动功能障碍。)。
这些结果表明,内皮细胞Mfn2缺失虽不能延长寿命,但可保护机体的多种生理、运动和行为参数免受衰老的影响。
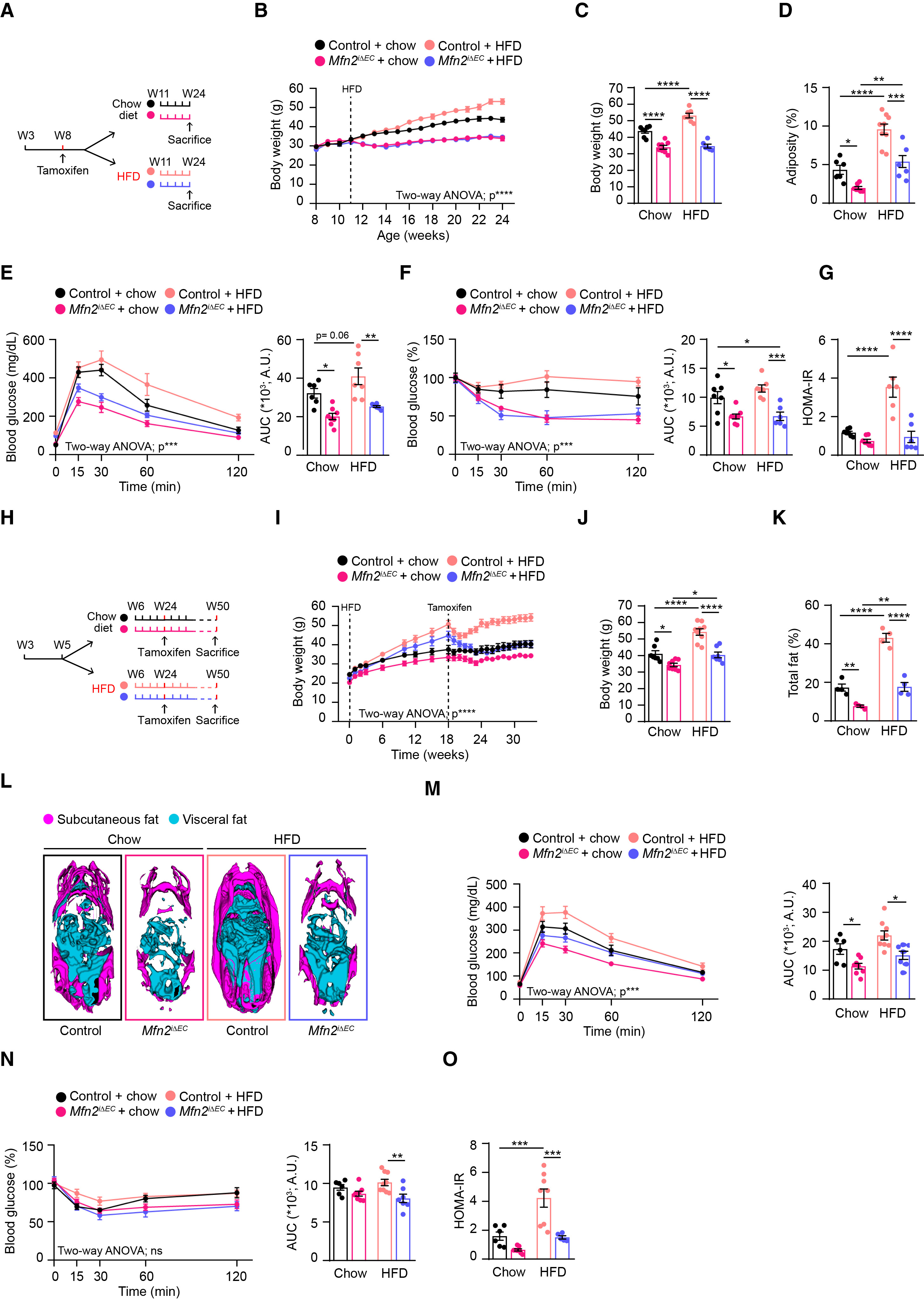 图6. 内皮细胞Mfn2的缺失可增强对饮食诱导肥胖的抵抗
图6. 内皮细胞Mfn2的缺失可增强对饮食诱导肥胖的抵抗
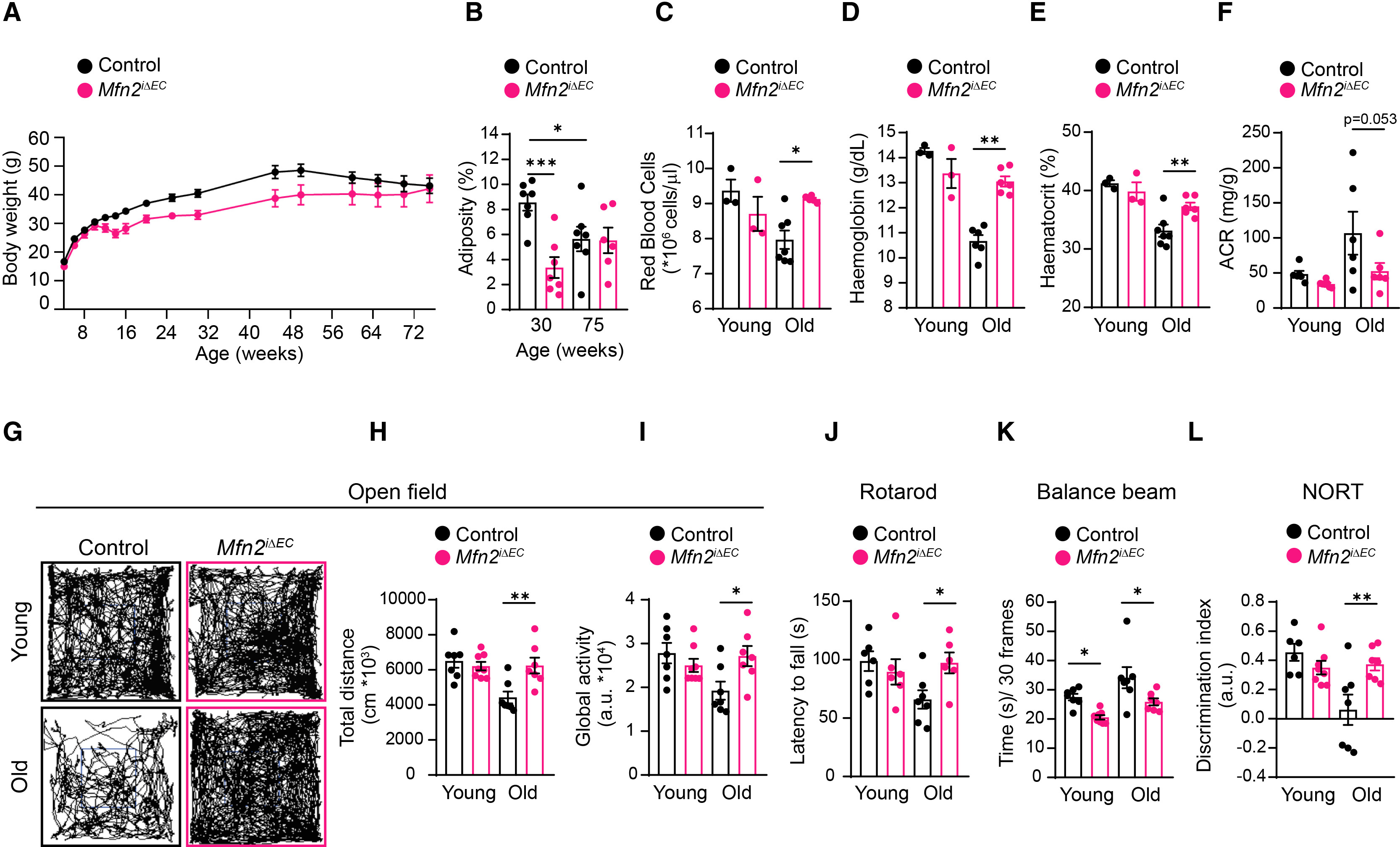
图7. 衰老Mfn2iΔEC小鼠健康状况改善
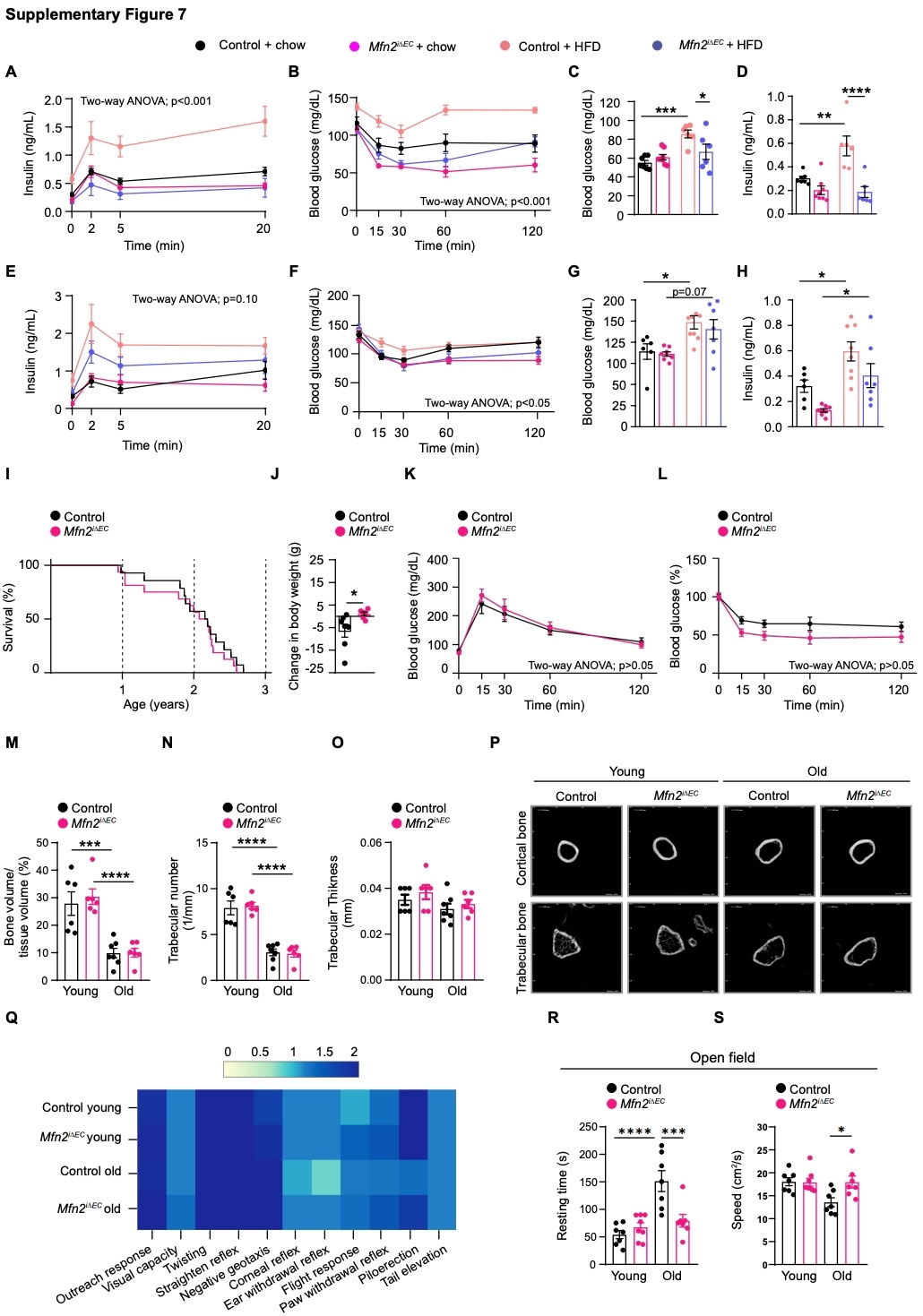
图S7. 关于接受高脂饮食或衰老的WT及Mfn2iΔEC小鼠表型特征的补充数据
总结
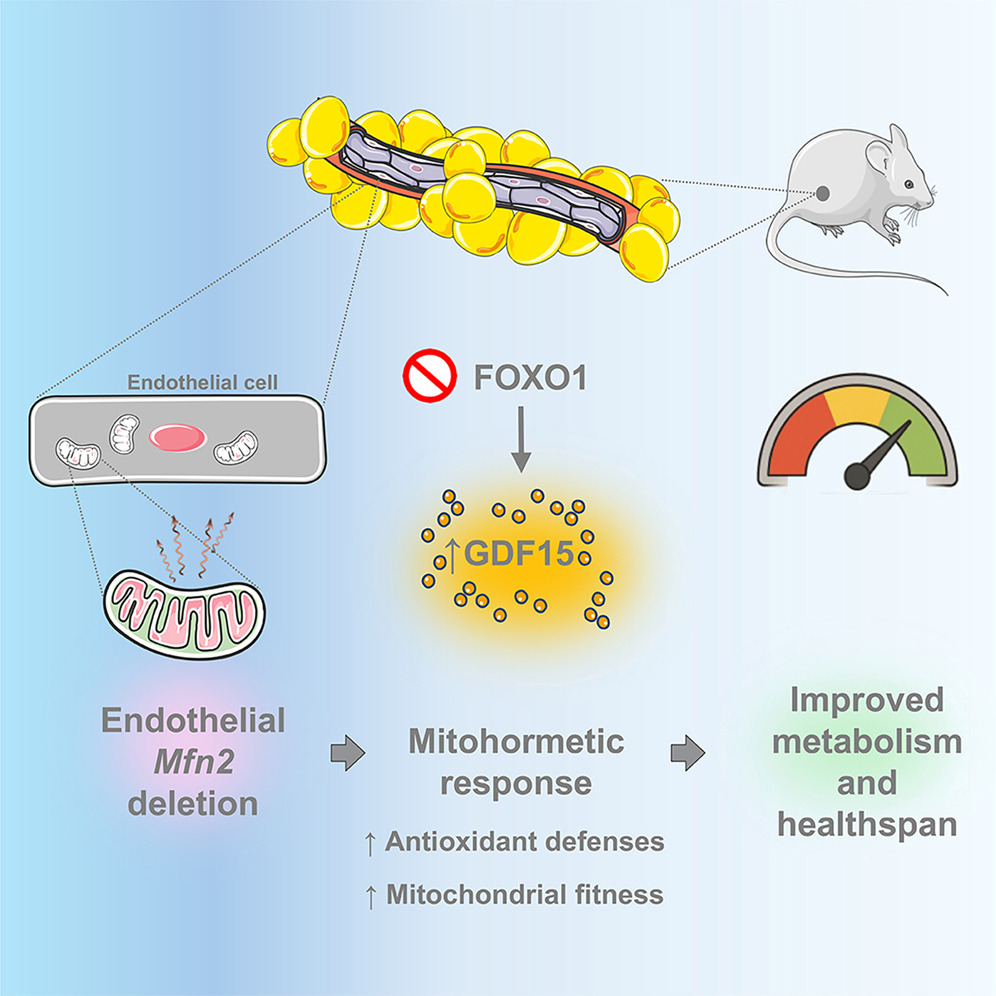
本研究揭示了,内皮细胞Mfn2缺失通过双重机制协同改善代谢稳态与衰老相关功能衰退:第一,激活内皮细胞线粒体低毒兴奋效应(mitohormesis),增强线粒体呼吸,促进脂解和脂肪酸氧化;第二,通过抑制FOXO1,来解除FOXO1对GDF15的转录抑制,促进内皮细胞分泌GDF15,进而激活最后区(AP)与孤束核(NTS)的GFRAL⁺神经元,抑制摄食并提升能量消耗,增强对饮食诱导肥胖的抵抗力,改善全身代谢与衰老相关表型。这一发现不仅重新定义了内皮细胞在代谢调控中的作用,也为靶向内皮细胞线粒体治疗肥胖与衰老相关功能障碍提供了新思路。
原文链接:https://www.cell.com/cell-metabolism/fulltext/S1550-4131(26)00012-4?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS1550413126000124%3Fshowall%3Dtrue
https://blog.sciencenet.cn/blog-3483272-1528787.html
上一篇:Science:线粒体,燃脂操盘手!
下一篇:Cell:老年痴呆这点事,肝说“酶”问题
