博文
浙大姜银珠等:氨基和羧基协同“锚定-捕获”构建锌负极稳定界面
|
研究背景
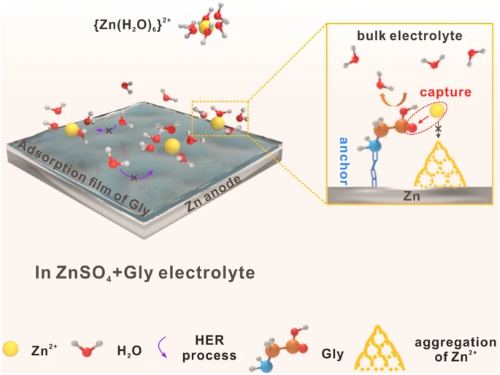
水系锌离子电池已被公认为是大规模储能应用中最具前景的电池体系之一,但锌负极-电解液界面的不稳定性极大地制约了其进一步发展。本文提出了一种基于甘氨酸(Gly)分子中氨基和羧基的协同“锚定-捕获”机制有效稳定负极的界面化学。通过同步耦合氨基在锌负极表面的锚定作用和羧基对局部区域锌离子的捕获作用,从而稳定负极-电解液动态界面,限制锌离子的无序迁移,并有效抑制副反应和枝晶生长。基于该机制作用,锌负极可逆性得到了显著提升,在68%的放电深度(DODZn)下实现长达200小时的稳定循环,获得了高性能水系锌离子电池。
Zhen Luo, Yufan Xia, Shuang Chen, Xingxing Wu, Ran Zeng, Xuan Zhang, Hongge Pan, Mi Yan, Tingting Shi*, Kai Tao, Ben Bin Xu* & Yinzhu Jiang*
本文亮点
内容简介
由于锌金属具有高理论容量(5855 mAh cm⁻³, 820 mAh g⁻¹)、低成本、高安全性和环境友好性等固有优点,水系锌离子电池正成为大规模储能系统的有力候选者。然而,枝晶生长和副反应这两个发生在负极-电解液界面且相互影响的问题严重阻碍了水系锌离子电池的进一步发展。浙大姜银珠&暨大时婷婷&诺桑比亚大学徐斌等人提出使用甘氨酸(Gly)作为电解液添加剂来解决上述问题。在循环过程中,由于氨基(-NH₂)与锌金属之间的化学相互作用,Gly分子可以稳定地锚定在锌负极表面,同时通过羧基(-COOH)在局部区域捕获锌离子,从而显著抑制枝晶生长和副反应。得益于这种协同“锚定-捕获”效应,组装的Zn-Zn对称电池在1 mA cm⁻²的电流密度下实现了超过2800 h的循环寿命。在高达68%放电深度下,依然能够稳定循环超200 h。Zn-MnO₂全电池在不同倍率下均表现出更高的放电容量和容量保持率。这项工作清楚地阐明了极性基团在抗腐蚀和锌离子电镀/剥离行为中的特定作用,为水系锌离子电池的电解液设计提供了新见解。
图文导读
I 氨基的“锚定”作用以抗腐蚀
为了准确比较不同极性基团的作用,选择N-乙酰甘氨酸(Ac-Gly)和甘氨酰胺盐酸盐(Gly-NH₂)作为另外两种添加剂,它们分别由甘氨酸中的氨基和羧基酰胺化衍生而来。Gly、Ac-Gly和Gly-NH₂的详细分子结构如图1a所示。在该工作中,所有的添加剂浓度均为0.1 M。如图1b的XRD图谱所示,锌片浸泡在加有三种添加剂的电解液中7天均未出现副产物的衍射峰。随后,Tafel和线性伏安扫描测试结果表明具有氨基的Gly和Gly-NH₂能进一步降低锌负极的腐蚀电流和析氢电位,有效阻碍副反应的发生(图1c, d)。而仅具有羧基的Ac-Gly只能在一定程度上降低析氢电流,对析氢电位影响不大,这可能来自于羧基的去溶剂化作用。为了理解锌负极在不同电解液中的抗腐蚀性能差异,进行了表面微分电容测试。如图1e所示,Gly和Gly-NH₂的引入显著降低了锌负极的表面微分电容,说明它们可以参与双电层结构并与锌负极表面相互作用。对浸泡后的锌片进行傅里叶红外光谱测试,出现-NH₂和C=O的伸缩峰证实了Gly和Gly-NH₂在锌负极表面的吸附作用(图1f, g)。图1h从理论计算的角度证实氨基比羧基具有更强的在锌表面的吸附作用,而图1i中的差分电荷密度结果表明Zn原子和N原子之间电子云出现重叠,说明是化学吸附。
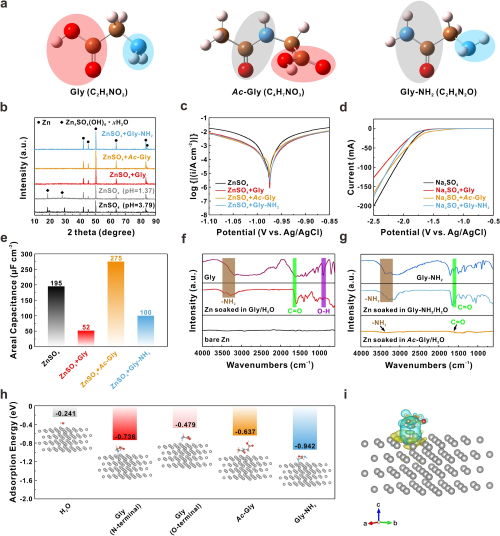
图1. (a) Gly、Ac-Gly和Gly-NH₂的分子结构;(b) 锌片在不同溶液中浸泡7天的XRD图谱;(c) 锌负极的Tafel曲线;(d) 锌负极的LSV曲线;(e) 锌在不同电解液中的平均差分电容;锌片在不同溶液中浸泡7天的FTIR光谱:(f) Gly/H₂O溶液;(g) Ac-Gly/H₂O和Gly-NH₂/H₂O溶液;(h) H₂O、Gly、Ac-Gly和Gly-NH₂分子在Zn(002)表面的吸附能比较;(i) Gly在Zn表面的差分电荷密度。
II 羧基的“捕获”作用以均匀锌沉积
如图2a所示,具有羧基的Gly和Ac-Gly引入后锌离子的形核过电位显著升高,远高于含Gly-NH₂电解液。计时电流测试结果表明,Ac-Gly和Gly-NH₂可以降低电流增长速率,这是由于锌离子的二维扩散受到限制所导致的。对于含Gly电解液,无序的锌离子通量被调节,锌离子直接在表面还原,形成稳定的三维扩散模式,电流密度响应最低(图2b)。随后,利用量子化学(QC)计算证明Gly和锌离子之间具有更强的结合能,同时分子动力学模拟(MD)结果表明Gly分子中羧基的双键氧原子参与到锌离子的溶剂鞘内,并替换一个水分子,从而改变了锌离子的溶剂化结构(图2c-g)。图2h的核磁共振图谱(NMR)中重水的²H峰从4.719 ppm偏移至4.716 ppm,进一步说明了Gly中羧基的去溶剂化作用。基于上述结果,Gly分子中氨基和羧基的协同“锚定-捕获”效应成为稳定锌负极的机制(图2i)。在ZnSO₄电解液中,锌离子与水分子溶剂化形成{Zn(H₂O)₆}²⁺,并在内电场作用下迁移至锌负极表面。一方面,水分子与金属锌接触后分解生成H₂,使局部区域pH值升高,进一步促进副产物(ZSH)的形成。另一方面,锌离子沿粗糙金属表面随机扩散,聚集在优先成核位置形成小凸起。连续恶化的表面条件显著诱导锌枝晶生长,最终刺穿隔膜造成短路,导致电池系统失效。与此相反,Gly分子的引入可以通过氨基的N原子牢固地锚定在锌负极表面,减少水分子与金属锌的接触,从而保护锌负极。同时,由于羧基的强亲核性,锌离子被Gly捕获,有效地限制了其二维扩散,并引导均匀沉积。对于Gly-NH₂和Ac-Gly而言,仅靠氨基或羧基不能有效地稳定界面,最终锌负极仍会恶化从而失效。
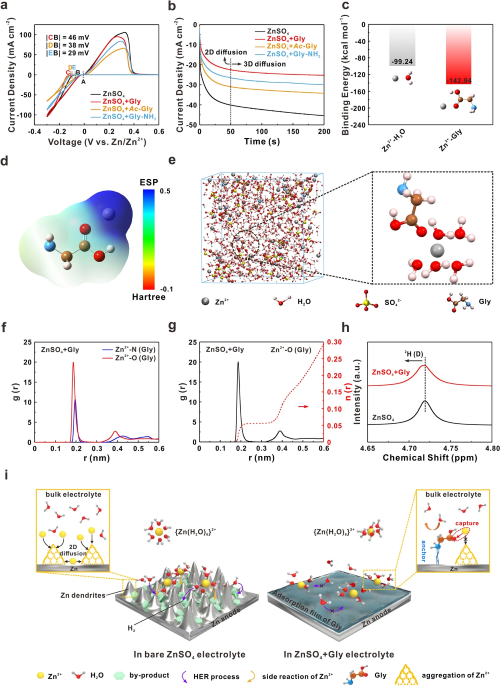
图2. (a) 锌在不同电解液中形核的CV曲线;(b) 锌负极的CA曲线;(c) 锌离子和不同分子(H₂O、Gly)之间的结合能;(d) Gly-Zn²⁺复合物的静电势;(e) MD模拟ZnSO₄ + Gly电解液的三维快照和锌离子溶剂化结构的局部放大快照;(f) Zn²⁺-N (Gly)和Zn²⁺-O (Gly)的模拟径向分布函数;(g) ZnSO₄ + Gly电解液中Zn²⁺-O (Gly)的模拟径向分布函数和配位数分析;(h) NMR图谱;(i) 锌沉积行为示意图。
III 协同“锚定-捕获”机制下的电化学性能
如图3a, b所示,Gly的引入使Zn-Ti非对称电池在1 mA cm⁻²和0.5 mAh cm⁻²条件下实现了超过500圈的超稳定循环,并表现出高达99.22%的平均库伦效率(CE),这说明锌负极在氨基和羧基的协同作用下可以保持高可逆性。对于ZnSO₄电解液,在第214次循环时,由于不断恶化的枝晶生长和严重的副反应,Zn-Ti电池的CE出现明显波动,无法达到充电截止电压(1.0 V)。而引入Ac-Gly和Gly-NH₂的Zn-Ti电池CE波动更为剧烈,主要是由于剥离不均匀以及在空穴外围或枝晶附近集中沉积所导致的。Zn-Zn对称电池在加入Gly后,在1或5 mA cm⁻²的电流密度下分别能够稳定循环超过2800 h和800 h,远远优于ZnSO₄电解液的性能(图3c, d)。图3e和f表明,Gly稳定负极-电解液界面后,锌倾向于平面沉积,大大降低了枝晶的形成概率,且在长期循环过程中界面能始终保持稳定不被破坏。另外,当锌利用率达到68%时,ZnSO₄电解液组装的对称电池仅能工作不到50 h,而Gly的协同机制可以实现长达200 h的稳定循环(图3g)。以上结果有力证明了协同“锚定-捕获”机制稳定锌负极的有效性。
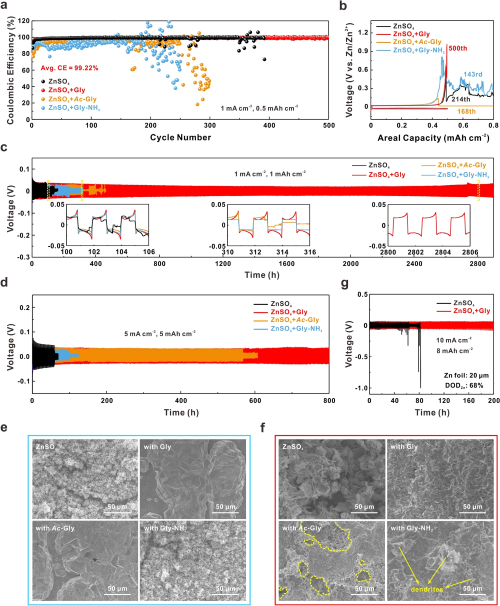
图3. (a) 不同电解液的Zn-Ti电池在1mA cm⁻²和0.5 mAh cm⁻²条件下的CE比较;(b) 对应不同循环圈数的容量-电压曲线;Zn-Zn对称电池在不同电流密度和面容量条件下循环的电压曲线:(c) 1 mA cm⁻², 1 mAh cm⁻²;(d) 5 mA cm⁻², 5 mAh cm⁻²;(e) 在5 mA cm⁻²下,Zn在不同电解液中电沉积1 h的SEM图像;(f) 在5 mA cm⁻²和5 mAh cm⁻²条件下,锌负极在不同电解质中循环50次后的表面形貌;(g) Zn-Zn对称电池在10 mA cm⁻², 8 mAh cm⁻²条件下循环的电压曲线(DODZn=68%)。
IV 稳定负极-电解液界面增强的MnO₂正极全电池验证
图4a展示了添加/不添加Gly的Zn-MnO₂全电池的CV曲线。两条曲线的氧化还原峰相似,说明Gly对MnO₂的反应机理没有影响。在ZnSO₄+Gly电解液中,阳极峰向较低电压移动(Δ₁=21 mV),阴极峰向较高电压移动(Δ₂=18 mV),表明MnO₂正极材料极化减弱,反应动力学提高。在协同“锚定-捕获”效应下,Zn-MnO₂全电池表现出比ZnSO₄电解液更好的倍率性能(图4b-d)。在经过0.2-1.0 A g⁻¹循环(每种电流密度循环5次)并回到0.2 A g⁻¹后,加入Gly的Zn-MnO₂电池的比容量恢复至309.4 mAh g⁻¹,其容量保持率高达82%,远高于ZnSO₄电解液。从图4e可以清楚地看到,在引入Gly的情况下,电池在200圈循环中保持稳定,在0.5 A g⁻¹电流密度下的放电比容量为167.2 mAh g⁻¹,而全电池在ZnSO₄电解液中循环80圈后容量快速衰减,在第200圈循环时,电池的放电容量仅为93 mAh g⁻¹(初始容量的31%)。以上结果证明了负极-电解液界面的稳定性在全电池中的重要作用。

图4. (a) Zn-MnO₂全电池的CV曲线;(b) Zn-MnO₂全电池在0.2-1.0 A g⁻¹电流密度下的倍率性能;(c)、(d) 对应充放电曲线;(e) 电流密度为0.5 A g⁻¹时Zn-MnO₂全电池的循环性能。
作者简介

本文通讯作者
▍主要研究成果
▍Email:ttshi@email.jnu.edu.cn

本文通讯作者
▍主要研究成果
▍Email:ben.xu@northumbria.ac.uk

本文通讯作者
▍主要研究成果
▍Email:yzjiang@zju.edu.cn
关于我们

E-mail: editor@nmlett.org
Tel: 021-34207624
https://blog.sciencenet.cn/blog-3411509-1402698.html
上一篇:NML文章集锦 | 钙钛矿材料研究论文(四)
下一篇:青岛大学吴广磊等:结构工程实现多重异质界面的集成,以获得轻质、柔韧和疏水的多功能电磁防护膜

全部作者的精选博文
全部作者的其他最新博文
- • 齐鲁工大李庆伟&河南大学张纪伟&香港城大Paul K. Chu等综述:钠离子电池硬碳材料构建“前-中-后”处理全流程
- • 中国科学院苏州纳米所吴晓东&天目湖先进储能院王志诚团队:稀释剂—阴离子解耦离子液体电解液,实现高压高安全锂金属电池
- • 东北大学青勇权: 超疏水可穿戴应变传感器:从设计到高稳定范式
- • 韩国全北国立大学Do Hwan Kim等:高极性掺杂CoFe-LDH用于海水电解双功能催化与防腐
- • 海工纪小宇&清华杨洋&中国科学院崔光磊等综述:聚氧化乙烯的分子内设计,解锁固态电解质与高能电池新蓝图
- • 过程所郭旸旸/姚明水/朱廷钰等综述:柔性MOFs在CO₂及其同位素中的吸附分离机制、调控策略与潜在应用