博文
SCRL:单细胞转录组数据的表示学习
||
SCRL:单细胞转录组数据的表示学习
单细胞转录组数据为我们进行细胞异质性的研究提供了强有力的工具,在处理这样一个高维的数据时,一个常用的策略是将这些细胞投影到低维的空间上。但是,与传统的转录组测序相比,单细胞测序技术噪声很大,使得单细胞转录组数据包含大量的dropout事件(导致基因表达量为0或接近0),即使是一些标记(marker)基因也有可能表达量很低。这样,一些传统的降维方法(比如在单细胞转录组数据分析中常常用到的PCA,t-SNE)就面临着巨大的挑战。为了克服这个问题,我们提出了一个基于网络嵌入(network embedding)的表示学习方法SCRL(见下图),通过数据驱动的非线性映射和引进先验知识(比如pathway information)来对细胞和基因学习一个更有意义的低维表示。同时SCRL对于异质性数据的整合提供了一种新思路。实验表明SCRL在多组近期的单细胞转录组数据上都表现卓越。
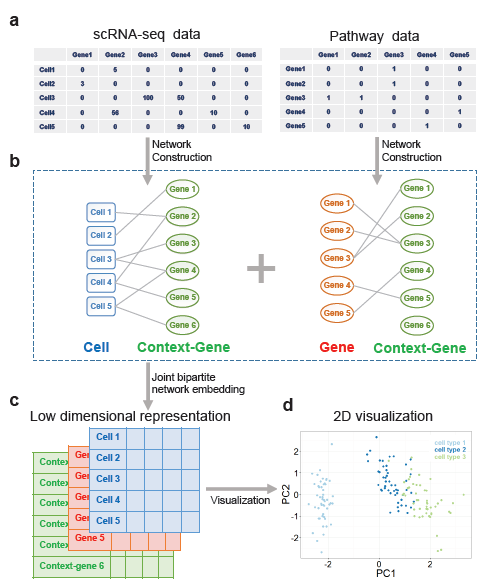
首先,我们从非监督学习(可视化/聚类)的角度出发,在三个数据集上进行了SCRL与PCA, t-SNE, ZIFA的性能比较,可视化结果(见下图)表明SCRL具有显著优势,特别是在Guo & Petropoulos数据集上。为了量化我们的实验结果,我们进一步计算了类内类间距离比(WB-ratio),其结果与可视化结果一致。

然后,我们从监督学习(分类)的角度出发,利用bootstrap的方法将数据分为训练集和测试集,在训练集上用不同的方法训练模型,并在测试集上计算正确率,结果(见下图)表明SCRL在Guo & Petropoulos数据集上具有显著优势,并且引入先验信息的分类正确率比不引入先验的分类正确率要高。在Pollen数据集上与其它方法效果相当。同时,我们在Guo数据集上利用我们对细胞和基因学习到的低维表示里找与不同的细胞类型对应的显著的pathway,结果与预期相符。此外,我们对PCA, t-SNE, ZIFA和SCRL在计算时间上进行比较,结果表明SCRL在大数据集上具有显著优势。

此项工作由清华大学古槿、张奇伟课题组联合完成,第一作者是清华大学自动化系博士生李翔宇,已被牛津出版社旗下著名期刊Nucleic Acids Research(影响影子10.162)接收。
X. Li, W.Chen, Y. Chen, X. Zhang, J. Gu* and M. Zhang*. Networkembedding basedrepresentation learning for single cell RNA-seq data. Nucleic Acids Research 2017, Advanced Access.
https://doi.org/10.1093/nar/gkx750
https://blog.sciencenet.cn/blog-407531-1071836.html
上一篇:斐特1区112(1~4)
下一篇:HCCDB--打造一站式检索的肝癌基因表达图谱