ВЉЮФ
ЪЙгУDiscovery StudioНјааздгЩФмМЦЫуНЬГЬЁЊЁЊFree Energy Perturbation
|
ФПЕФЃКВЩгУDiscovery StudioЃЌвдвЛзщаЁЗжзгЛЏКЯЮяЮЊЪЕР§ЃЌЪОЗЖFEPМЦЫуМАНсЙћЗжЮіВйзїЙ§ГЬЁЃ
ЫљашЙІФмЃКDiscovery Studio clientЃЌDS Preparing analogs and ligand pairsЁЂDS Setting up the relative FEP calculationsЁЂDS Running the FEP calculationsЁЂDS Collating and analyzing the FEP results
ЫљашЮФМўЃК4YKR_prep.dsvЁЂhsp90_analogs.sdЁЂhsp90_lead.sd
ЫљашЪБМфЃК30Зжжг+FEPМЦЫуЪБМфЃЈгыЕчФдгВМўЯрЙиЃЉ
НщЩмЃКРэНтКЭСПЛЏХфЬхКЭЕААзжЪжЎМфЯрЛЅзїгУЧПЖШЕФФмСІдквЉЮяЗЂЯжЯюФПжаЪЧжСЙиживЊЕФЃЌвдЪЖБ№ХфЬхЕФБфЛЏШчКЮПЩФмИФБфЧзКЭСІЁЃЖдНсКЯЧзКЭЖШЕФПЩППЙРМЦПЩвдИФНјЯШЕМгХЛЏЙ§ГЬжаХфЬхЕФбЁдёЁЃдкдчЦкЯШЕМгХЛЏНзЖЮЃЌПЩФмЛсПМТЧГЩЧЇЩЯЭђжжВЛЭЌжЇМмМвзхЕФХфЬхЃЌвђДЫжюШчЖдНгКЭMM-GBSAЕШМЦЫуЗНЗЈЪЪгУгкЫљашЕФЭЬЭТСПЁЃКѓЦкЯШЕМгХЛЏЩцМАаЁЕФЁЂЫГађЕФЛЏбЇаоЪЮЃЌашвЊИќОЋШЗЃЌПЩФмашвЊУїШЗЕФШмМСНЈФЃКЭбЯИёЕФздгЩФмЗНЗЈЁЃСЖН№ЪѕздгЩФмЩуЖЏ(FEP)ЪЧНЈСЂдкШШСІбЇбЛЗЛљДЁЩЯЕФЁЃ
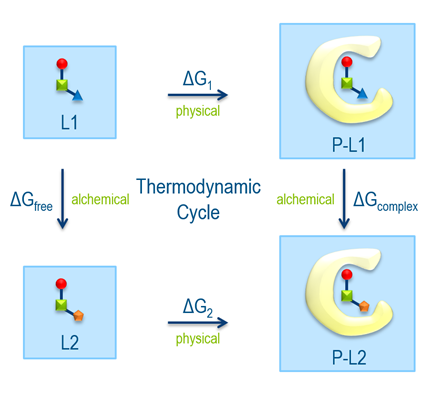
ЫЎЦНЮяРэЙ§ГЬУшЪіСЫЕААзжЪИДКЯЮяжаДгШмМСжаЮДНсКЯЕФХфЬхЕННсКЯЬЌЕФздгЩФмБфЛЏЁЃдкДЙжБСЖН№ЪѕЙ§ГЬжаЃЌХфЬх1дкЗЧНсКЯЬЌКЭНсКЯЬЌзЊБфЮЊХфЬх2ЁЃетЪЧЭЈЙ§вЛЯЕСаЗЧЮяРэжаМфзДЬЌНјааМЦЫуЕФЁЃІЄGcomplex - ІЄGfreeЕФСЖН№ЪѕЯрЖдЪјИПздгЩФмЕФМЦЫуЯрЕБгкІЄG2 - ІЄG1ЃЌЕЋМЦЫуГЩБОНЯЕЭЁЃ
БОНЬГЬжївЊАќРЈвдЯТВНжшЃК
жЦБИРрЫЦЮяКЭХфЬхЖд
НЈСЂЯрЖдFEPМЦЫу
дЫааFEPМЦЫу
ећРэКЭЗжЮіFEPНсЙћ
вЛЁЂжЦБИРрЫЦЮяКЭХфЬхЖд
FEPЖЏСІбЇФЃФташвЊЫљгаИааЫШЄЕФХфЬхЖЈЮЛдкЕААзжЪЛюадЮЛЕуЩЯЁЃдкЕквЛВНжаЃЌФњНЋЪЙгУЩњГЩФЃФтЙЙЯѓавщНЋФњЕФХфЬхгыЕААзжЪЛюадЮЛЕужаЕФЯШЕМХфЬхЖдЦыЁЃИУавщЭЈЙ§зюДѓЙЋЙВзгНсЙЙ(MCS)ЗНЗЈМьВтУПИіФЃФтЮяКЭЯШЕМЛЏКЯЮяжЎМфЕФЙЋЙВдзгКЭВЛЭЌдзгЁЃЫќжБНгДгЯргІЕФЧІдзгЗжХфФЃФтЮяЕФЙЋЙВдзгЕФзјБъЃЌВЂЮЊВЛЭЌЕФдзгЩњГЩаТЕФзјБъЁЃ
ШШанПЫЕААз90ЕААзХфЬхИДКЯЬхЕФPDBБрТыЮЊ4YKRЁЃБОНЬГЬжаЪЙгУЕФЪОР§ЮФМўЪЧДгPDBЭјеОЯТдиЕФЃЌЕААзжЪЪЧгУPrepare ProteinsавщжЦБИЕФЁЃдкжЦБИЕААзжЪЪБЃЌвЊЬиБ№зЂвтОЇЬхЫЎЃЌвдШЗЖЈЫќУЧЪЧЗёгІИУзїЮЊБЃЪиЕФЫЎЗжзгБЃСєЁЃЩюТёдкЕААзжЪКЫаФЕФЫЎЗжзгПЩФмдкЮЌГжЕААзжЪЕФелЕўНсЙЙжаЗЂЛгзїгУЃЌВЂОГЃгыМЋадЛљЭХаЮГЩЧПЧтМќЁЃдкБОНЬГЬжаЃЌЮвУЧдкНсКЯЮЛЕуБЃСєСЫШ§ИівбжЊЕФгыЯШЕМЛЏКЯЮяаЮГЩживЊЯрЛЅзїгУЕФЫЎЗжзгЃЌВЂдкећИіЙЄзїСїГЬжаЪЙгУЯрЭЌЕФЕААзжЪНсЙЙЁЃШЛЖјЃЌдкЮДРДЕФгІгУжаЃЌЬиЖЈЕФЫЎЗжзгЕФЯрЙиадзюГѕПЩФмВЛжЊЕРЃЌНЈвщЩњГЩФЃФтЙЙЯѓавщдЫааЪБЃЌЕААзжЪжаВЛДцдкШЮКЮОЇЬхЫЎЗжзгЃЌFEPЩшжУгыЕААзжЪНсЙЙБЃСєЫљгаЕФЫЎЗжзгЁЃ
1. ДђПЊ4YKR_prep.dsvЁЂhsp90_analogs.sdЁЂhsp90_lead.sdетШ§ИіЮФМўЃЌЮЛгкSamples | Tutorials | Simulation |ЮФМўМаЁЃ
2. Generate Analog ConformationsЃЌИУГЬађИљОнвЛЯЕСаЯШЕМЛЏКЯЮяЕФвЛИіЛђЖрИіЯШЕМЛЏКЯЮяЕФШ§ЮЌМИКЮНсЙЙКЭЯргІЕФФПБъЕААзжЪНсЙЙЃЌдЪаэЛёЕУЭЌаЭФЃФтЛЏКЯЮяЯЕСажаХфЬхЕФШ§ЮЌМИКЮНсЙЙЁЃИУавщЪзЯШЭЈЙ§зюДѓЙЋЙВзгНсЙЙ(MCS)ЗНЗЈМьВтУПИіФЃФтЮяКЭЯШЕМЛЏКЯЮяжЎМфЕФЙЋЙВдзгКЭВЛЭЌдзгЁЃЫќжБНгДгЯргІЕФЧІдзгЗжХфФЃФтЮяЕФЙЋЙВдзгЕФзјБъЃЌВЂЩњГЩВЛЭЌдзгЕФзјБъЁЃгЩгкФЃФтЛЏКЯЮяПЩвдгаВЛжЙвЛжжЗНЪНгыЯШЕМЛЏКЯЮяЭиЦЫЦЅХфЃЌЖјЧвгЩгкВЛЭЌЕФдзгЛсЩњГЩЖрИіЙЙЯѓЃЌвђДЫЪфГіЭЈГЃЛсЕМжТУПИіФЃФтЛЏКЯЮяВњЩњЖрИіЙЙЯѓЁЃШчЙћЬсЙЉСЫЕААзжЪЪмЬхЃЌЖдгкУПвЛИіЙЙЯѓЃЌЭЈЙ§ЖдИДКЯЬхКЭздгЩЕААзжЪКЭХфЬхНјааЕЅЕуCHARMmФмСПМЦЫуЕУЕННсКЯФмЃЌВЂвдФмСПзюгаРћЕФЙЙЯѓгХЯШХХађЁЃШЛЖјЃЌетжжФмСПВЛЬЋПЩФмПЩППЕидЄВтНсКЯЧзКЭадЃЌвђЮЊЫќВЛЩцМАНсЙЙгХЛЏЁЃДђПЊGenerate Analog ConformationsАДееЯТЭМНјааВЮЪ§ЩшжУЃЌдЫааЁЃ
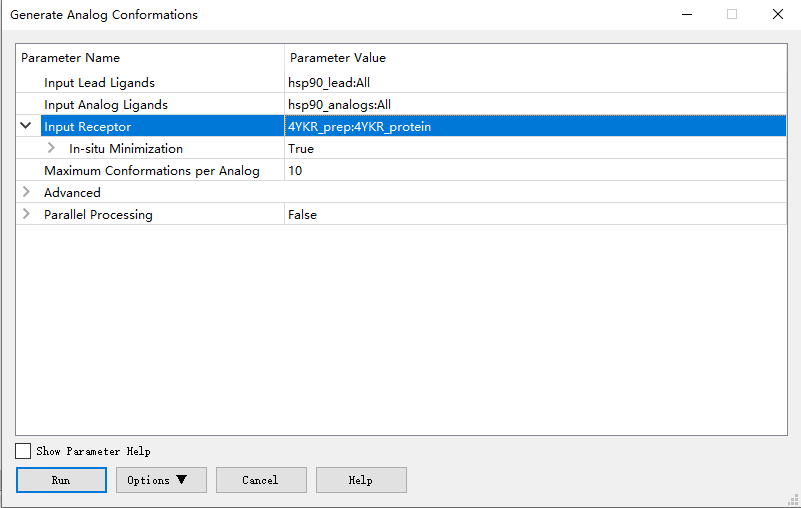
3. НсЙћЮФМўжаAnalog ConformationsгЕга8ИіЙЙЯѓЃЌЦфжаhsp90_52гаСНИіЙЙЯѓЃЌTop Scoring ConformationsЮФМўздЖЏМЏКЯКЭДђЗжзюИпЕФУПИіХфЬхЕФЙЙЯѓЃЌвђДЫбЁдёетИіЮФМўМДПЩЁЃИјИУ7ИіаЁЗжзгЬэМгСІГЁЁЃSimulation | Change ForcefieldЃЌбЁдёCHARMmСІГЁЃЌApply ForcefieldгІгУСІГЁЃЈFEPЫуЗЈЭЌЪБжЇГжCHARMmКЭcharmm36СНжжСІГЁЃЌШєвЊЪЙгУcharmm36дђгУType Ligands with MATCH (Prototype)ГЬађгІгУЃЉЁЃ
4. ДђПЊGenerate Ligand Pairs for FEP....ЃЌИУГЬађЕФЙЄзїдРэЪЧЪзЯШЮЊУПИіЮЈвЛЕФХфЖдМЦЫувЛИіАќКЌзюДѓЙЋЙВзгНсЙЙ(MCS)ЕФЯрЫЦадОиеѓЁЃДгетИіОиеѓжабЁдёХфЬхжЎМфЕФСЌНгЃЌЪЙДѓЖрЪ§ЯрЫЦЕФХфЬхСЌНгЕНПЩФмЕФзюМбЗЖЮЇЃЌетЪЧЪЙгУзюДѓЯрЫЦЖШЫуЗЈЭъГЩЕФЁЃЖдгкКЌгаNИіХфЬхЕФЪ§ОнМЏЃЌНсЙћЪЧN-1ЖдЕФМЏКЯЁЃ АДееШчЯТЩшжУЃЌвђЮЊЕк3ВНЕФhsp_90 leadЮФМўОЭЪЧhsp_12ЃЌвђДЫНЋдкTop Scoring ConformationsЮФМўРяЕФhsp_12бЁдёЮЊЯШЕМХфЬхЃЈПЩвдздаажИЖЈЃЉЃЌдЫааЁЃ
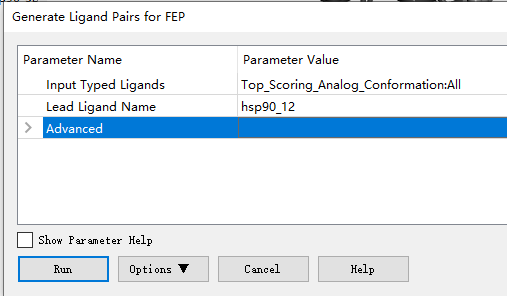
5. дЫааНсЙћШчЯТЭМЫљЪОЃЌ

ЗжзгДАПкжаЕФХфЬхЖдБЛЖЈвхЮЊЕквЛИіХфЬхЭЈЙ§СЖН№ЪѕзЊБфЮЊЕкЖўИіХфЬхЕФзщКЯЁЃЭјТчЭМЯдЪОСЫХфЬхЖджЎМфЕФЙиЯЕЃЌЦфжаНкЕуЪЧХфЬхЃЌБпБэЪОХфЖдЁЃБпдЕБЛБъМЧЮЊвЛЖдЕФЯрЫЦЖШЗжЪ§ЃЌМ§ЭЗБэЪОСЖН№ЪѕБфЛЏЕФЗНЯђЁЃФуПЩвдбЁдёжИЖЈвЛИіЯШЕМХфЬхЃЌдкетжжЧщПіЯТЃЌУПЖдХфЬхИљОнЫќУЧгыЯШЕМХфЬхЕФОрРыХХСаЃЌЦфжаОрРыЪЧгЩХфЬхКЭЯШЕМХфЬхжЎМфЕФНщШыНкЕуЕФЪ§СПРДКтСПЕФЁЃhsp90_12ХфЬхгыдк4YKRжаНсОЇЕФХфЬхЯрЭЌЁЃPDBИДдгЃЌгаСЫЭјТчЭМЃЌФуПЩвдДгЯШЕМПЊЪМЃЌЯђЭтЕЅЯђвЦЖЏЕНдНРДдНВЛЯрЫЦЕФРрЫЦЮяЁЃ
6. НгзХЮвУЧашвЊзМБИдЫааFEPМЦЫуЕФНсЙЙЁЃИљОндкЭиЦЫКЭЗЧМќВЮЪ§жаЪЖБ№ГіЕФЙВгаКЭВЛЭЌдзгМЏЃЌУПИіХфЬхЖдЩњГЩКЯВЂЕФЖдХМЭиЦЫЗжзгЁЃЛЙЩњГЩСЫетжжКЯВЂЕФЖдХМЭиЦЫЗжзгЕФШмМСЛЏХфЬхЬхЯЕКЭШмМСЛЏЕААзжЪХфЬхТчКЯЮяЁЃДђПЊSet Up Relative FEP CalculationsЁАДееШчЯТНјааЩшжУЃЌЮвУЧПЩвдЩшжУжИЖЈЕНжмЦкадБпНчЬѕМўЕФзюДѓОрРывдМАcell shapeаЮзДЁЂЪЧЗёЬэМгбЮШмвКЛЗОГЕШЃЌдЫааЁЃ
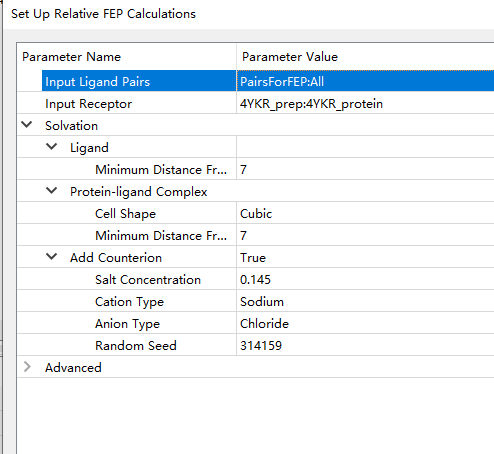
7. дкСЖН№ЪѕFEPМЦЫужаЃЌРћгУКЯВЂЕФЖдХМЭиЦЫЗжзгНЋвЛИіХфЬхзЊЛЏЮЊЕкЖўИіХфЬхЁЃетжжЧЖКЯЗжзгЪЧЭЈЙ§зюДѓЙВзгНсЙЙ(MCS)ЗНЗЈЪзЯШМьВтХфЬхжЎМфЕФЙВгадзгКЭВЛЭЌдзгЖјВњЩњЕФЁЃЦеЭЈдзгЪЧФЧаЉдкЭиЦЫНсЙЙЁЂдзгРраЭКЭВПЗжЕчКЩЗНУцБЛШЯЮЊЪЧЯрЭЌЕФдзгЁЃЫљгаЦфЫћдзгЖМБЛжИЖЈЮЊВЛЭЌЕФдзгзщЁЃдЫааНсЙћШчЯТЃЌЦфжавЛИіАќКЌКЯВЂЕФЖдХМЭиЦЫЗжзгдквЛИіСЂЗНаЮзДЕФКазгРяЃЌЫЎЗжзгКЭбЮРызгЁЃСэвЛИіЗжзгДАПкАќКЌСЫУПИіЖдХМЭиЦЫЗжзггыЕААзжЪдкРрЫЦШмМСЛЏКазгжаЕФИДКЯЮяЁЃ
8. ЗжНчЕуЁЃЧАЦкЕФЮФМўзМБИЙЄзїЕНДЫНсЪјЁЃе§ЪНЕФFEPМЦЫудкDSжагаCPUКЭGPUЃЈНижЙАцБОЮЊDS2022ЃЌднЪБжЛжЇГжLinuxЯЕЭГЃЉСНжжЗНЪНЁЃЯТУцЗжБ№еЙЪОЁЃ
9.1. CPUМЦЫуЁЃдЫааСНБщFree Energy Perturbation (FEP)ГЬађЃЌвЛБщЮЊМЦЫуШмМСЛЏЪмЬхЃЌвЛБщМЦЫуШмМСЛЏХфЬхЃЌШчЯТЭМЫљЪОЁЃЮвУЧПЩвддкетРяШЅаоИФвЛаЉВЮЪ§жюШчLambdaДАПкКЭЩњВњЕФЪБМфВНГЄЕШвдЪЪгІВЛЭЌЕФМЦЫуашЧѓЁЃ
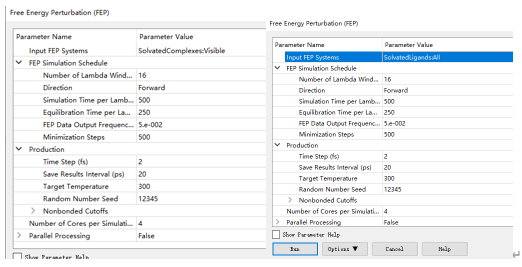
дЫааНсЙћШчЯТЃК

ЦфжавЛИіFEPНсЙћБЈИцЁЃ
10.1 ећРэКЭЗжЮіFEPНсЙћЃЌдкзЪдДЙмРэЦїжаДђПЊЩЯЪіНсЙћЮФМўЃЈвВПЩвддкjobжагвМќlocateЖЈЮЛЃЉЃЌЦфжаАќКЌвЛаЉЮФМўЃЌШчЯТЁЃ

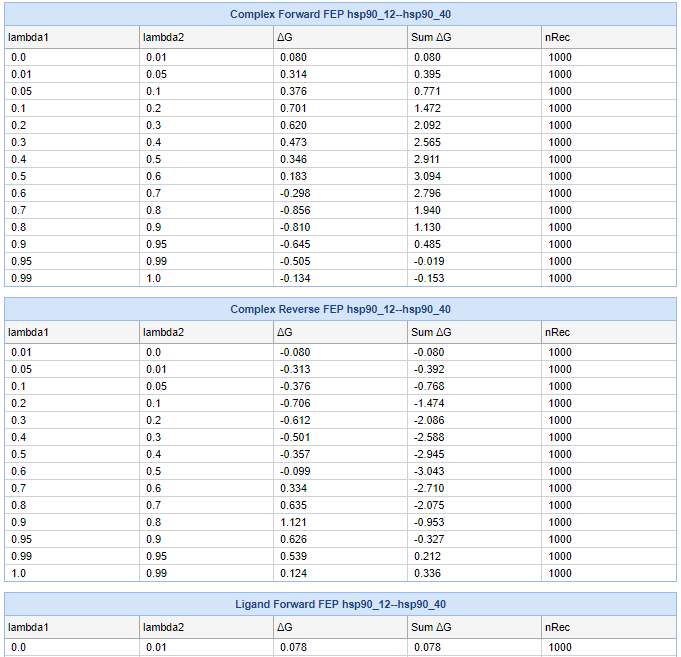
етаЉЮФМўМажаЕФНсЙћАќКЌУПИіХфЬхЖдЕФвЛИіsimulationinfo.xmlЮФМўКЭвЛИіздгЩФмЪ§ОнЮФМў(.fepout)ЁЃxmlЮФМўжИЖЈСЫУПИіШХЖЏжаЩцМАЕФХфЬхЕФУћГЦЃЌвдМАФЃФтЪЧЩцМАздгЩХфЬхЛЙЪЧИДХфЬхЁЃ.fepoutЮФМўЪЧдкNAMD FEPЩњВњдЫааЦкМфЩњГЩЕФЃЌЦфжаАќКЌдЪМздгЩФмЪ§ОнЁЃ
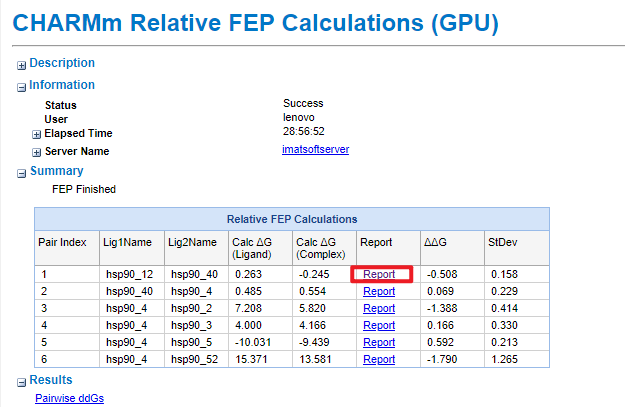
ДђПЊCollate FEP Simulation Results....ГЬађЃЌНЋFEPЪ§ОнФПТМХфЬхВЮЪ§ЩшжУЮЊШмМСХфЬхЩЯNAMD FEPМЦЫуЕФНсЙћЮЛжУЃЌНЋFEPЪ§ОнФПТМИДКЯЬхВЮЪ§ЩшжУЮЊШмМСИДКЯЬхЩЯNAMD FEPМЦЫуЕФНсЙћЮЛжУЁЃЃЈШчЙћашвЊМЦЫуОјЖдздгЩФмЃЌдђеЙПЊОјЖдздгЩФмВЮЪ§зщЃЌВЂдкЯШЕМХфЬхУћГЦВЮЪ§жаЪфШыhsp90_12ЁЃдкЯШЕМХфЬхздгЩФмВЮЪ§зжЖЮжаЪфШыжЕ-10.27ЃЈЪЕбщжЕЃЉЁЃЩшжУЪфШыХфЬхВЮЪ§ЮЊTop_Scoring_Analog_Conformation:All.ЃЉЃЌдЫааЁЃ
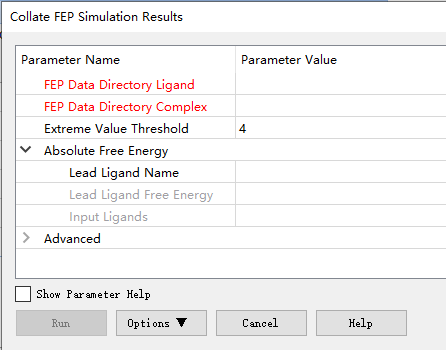
дЫааНсЙћШчЯТЫљЪОЃЌСаГівЛИіМЦЫуГіЕФХфЬхЖдЕФЯрЖдздгЩФмжЕЕФБэЃЌвдМАвЛИіХфЬхЕФОјЖдздгЩФмБэЁЃКѓвЛИіБэжЛгадкФуЬсЙЉСЫЯШЕМХфЬхКЭздгЩФмжЕЪБВХЛсЩњГЩЁЃетСНИіБэвВЖМгУжљзДЭМЭМаЮЛЏБэЪОЁЃ
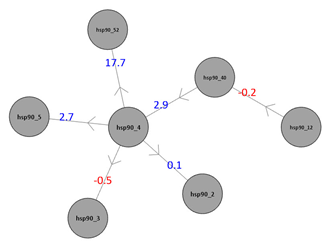
ФуЛсзЂвтЕНЃЌhsp90_52ЕФОјЖдздгЩФмдкБэИёжаБЛБъМЧЮЊЁАПЩФмВЛПЩППЁБЃЌдкЭМБэжаБЛБъМЧЮЊКьЩЋЁЃетЪЧвђЮЊОбщЙлВьБэУїЃЌдкЭЌЪєЪ§СажаЃЌЯрЖдздгЩФмжЕЕФДѓаЁКмЩйДѓгк3ЧЇПЈ/ФІЖћЁЃМЋжЕуажЕВЮЪ§дЪаэФњБъМЧетаЉПЩФмВЛПЩППЕФдЄВтЃЌетПЩФмЪЧгЩгкФЃФтжаЕФВЛзМШЗадЃЌШчгЩгкВЩбљВЛзуЛђСІГЁВЛЪеСВЁЃЖдгкФГаЉАќКЌЗЧЛюадЗжзгЕФЪ§ОнМЏЃЌ3ЧЇПЈ/ФІЖћЕФуажЕПЩФмБъжОзХДѓСПЕФдЄВтЁЃдкетжжЧщПіЯТЃЌЭЈЙ§НЋуажЕЬсИпЕН4Лђ5ЧЇПЈ/ФІЖћРДВтЪдНсЙћЖдуажЕЕФУєИаадЪЧгагУЕФЁЃ
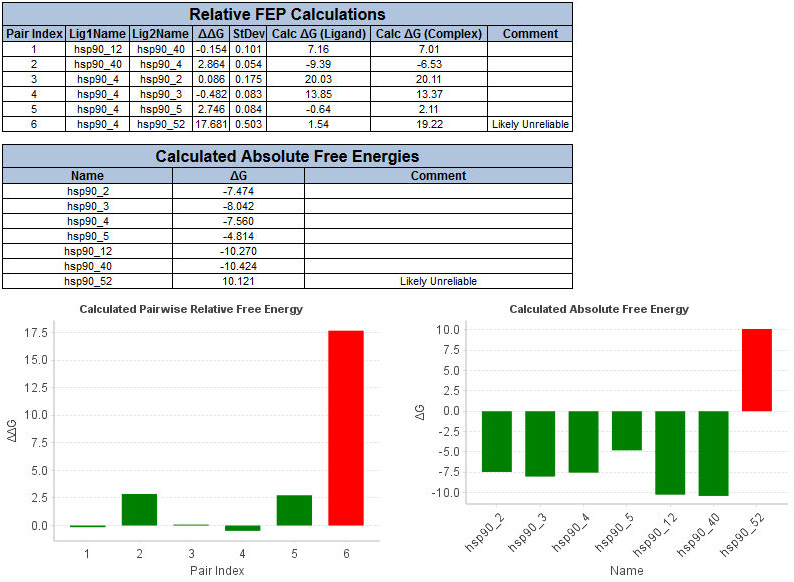
ШчЙћФуНЋетаЉжЕгыБЈИцжаМЦЫуГіЕФОјЖдІЄGжЕНјааБШНЯЃЌФуЛсЗЂЯжЃЌГ§СЫВЛПЩППЕФhsp90_52дЄВтЭтЃЌздгЩФмЩуЖЏМЦЫуФмЙЛКЯРэЕидЄВтОјЖджЕЁЃ
вВаэБШжиЯжОЋШЗЕФОјЖдздгЩФмдЄВтИќживЊЕФЪЧЃЌFEPЗНЗЈФмЙЛЖдХфЬхНјааХХађЁЃЕБОјЖдздгЩФмдЄВтВЛФмМЦЫуГіХХађЪБЃЌФуШдШЛПЩвдЪЙгУЯрЖджЕНјааЗжЮіЁЃПДПДХфЬхЖдhsp90_40 - hsp90_4ЕФІЄІЄGжЕЮЊ2.864ЃЌПДПДhsp90_4 - hsp90_3ЕФжЕЮЊ-0.482ЁЃФуПЩвдОнДЫЭЦЖЯЃЌhsp90_40ЕФНсКЯФмБШhsp90_4КУЃЌЖјhsp90_3ЕФНсКЯФмБШhsp90_4КУЁЃХфЬхЖдhsp90_4 - hsp90_5ЕФІЄІЄGжЕЮЊ2.746ЃЌЫЕУїhsp90_5ЕФНсКЯФмзюВюЁЃ
Collate FEPФЃФтНсЙћавщЛЙЩњГЩвЛИіЭјТчЭМ(ШчЭМ)ЃЌЭМаЮЛЏЕиБэЪОХфЬхЖдЕФІЄІЄGжЕЁЃБпдЕгУХфЬхЖдЕФЯрЖдздгЩФмНјааБъМЧКЭбеЩЋБрТыЃЌМ§ЭЗБэЪОСЖН№ЪѕБфЛЏЕФЗНЯђЁЃ
дкБОНЬГЬжаЃЌФњвбОЪЙгУСЫЯрЖдСЖН№ЪѕздгЩФмШХЖЏРДЮЊКѓЦкЯШЕМгХЛЏЕФвЛЯЕСаЭЌаЭХфЬхХХађЁЃФЃФтЫљашЕФМЦЫуГЩБОЯожЦСЫИУЗНЗЈЕФаЁЪ§ОнМЏЃЌЕЋЦфЬсЙЉИќОЋШЗЕФНсКЯФмЖЈСПЙРМЦЕФЧБСІЪЙЦфГЩЮЊЛљгкКЯРэНсЙЙЕФвЉЮяЩшМЦЕФБІЙѓЙЄОпЁЃ
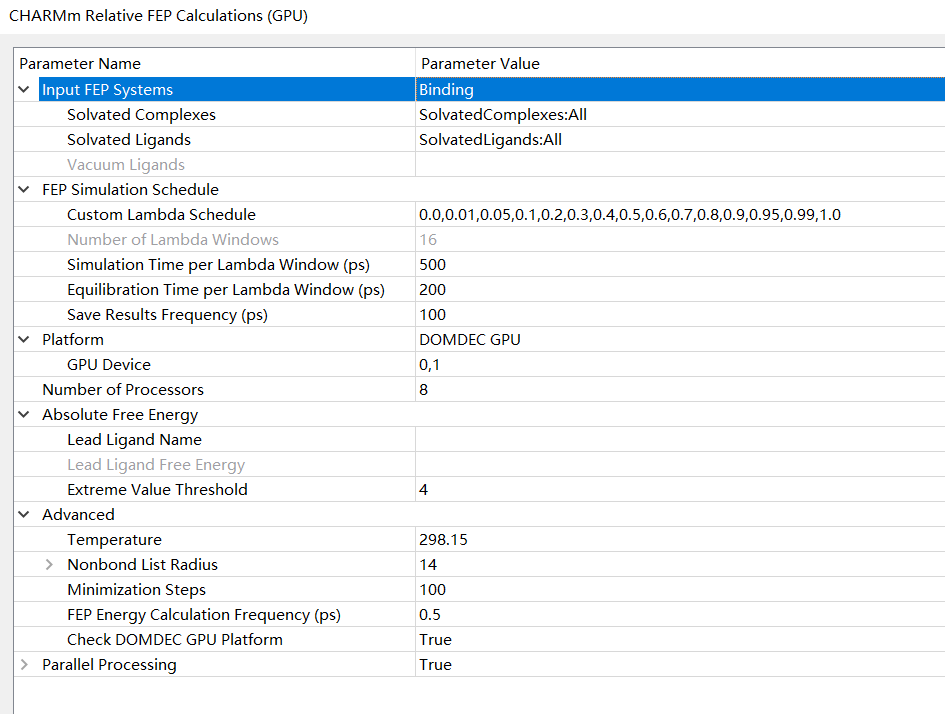
9.2 GPUМЦЫуЁЃGPUМЦЫуФПЧАНіФмдкLinuxЯЕЭГЃЈDSЗўЮёЦїАВзАдкLinuxЯЕЭГЃЌDSПЭЛЇЖЫвРОЩПЩвдАВзАдкwindowsЩЯЃЉЩЯдЫааЃЌwindowsЯЕЭГднЪБВЛжЇГжЁЃGPUМЦЫуећКЯСЫЗжЮіНсЙћЃЌПЩвдвЛВНЩњГЩНсЙћЁЃДђПЊCHARMm Relative FEP Calculations (GPU)ГЬађЃЌВЮЪ§ШчЯТЁЃGPUГЬађЯТЃЌЮвУЧНіашХмвЛДЮГЬађМДПЩЃЌдкетРяПЩвдШчCPUГЬађвЛбљаоИФвЛаЉВЮЪ§жюШчLambdaДАПкКЭЩњВњЕФЪБМфВНГЄЕШвдЪЪгІВЛЭЌЕФМЦЫуашЧѓЁЃЭЌЪБШчЙћашвЊМЦЫуОјЖдздгЩФмЃЌдђбЁдёЯШЕМХфЬхЃЌЬюаДЪЕбщжЕМДПЩЁЃ
НсЙћШчЯТЃК
жЕЕУзЂвтЕФЪЧЃЌGPUдЫааFEPГЬађгаШ§жжFEP SystemsПЩЙЉЪфШыЃЌЗжБ№ЪЧBindingЃЈашвЊШмМСЛЏЕФИДКЯЮяКЭШмМСЛЏЕФХфЬхзїЮЊЪфШыЃЉЁЂHydrationЃЈашвЊШмМСЛЏХфЬхКЭецПеХфЬхзїЮЊЪфШыЃЌПЩвдНсЫуЫЎКЯздгЩФмЃЉЁЂSingleЃЈдЫааШ§ИіПЩФмЕФЯЕЭГжаЕФвЛИіЃЌдкдЫааСНИіЕЅвЛРраЭЕФМЦЫуКѓЃЌЪжЖЏНсКЯНсЙћЃЌвдЛёЕУBindingЛђHydrationздгЩФмЃЉЁЃЦфжаецПеХфЬхЕФЮФМўПЩдкSet Up Relative FEP CalculationsЕФЪфГіНсЙћжаЛёЕУЁЃ

https://blog.sciencenet.cn/blog-3536821-1362724.html
ЩЯвЛЦЊЃКDiscovery Studio | Multi-Site Lambda Dynamics (MSLD) НјааздгЩФмМЦЫу
ЯТвЛЦЊЃКЁОзджїбаЗЂЁПШчКЮЪЙгУDiscovery StudioНјааЗДЯђевАа