博文
Cell:肝脏肥胖求生 为肝癌悄悄踩油门
||
代谢学人
Cell:肝脏肥胖求生 为肝癌悄悄踩油门
撰文 | 陈明洁 皮婉莹 李姿萱 郭钰涵 周文豪 邱瑾
编辑 | 孟美瑶
校对 | 陈明洁

背景介绍
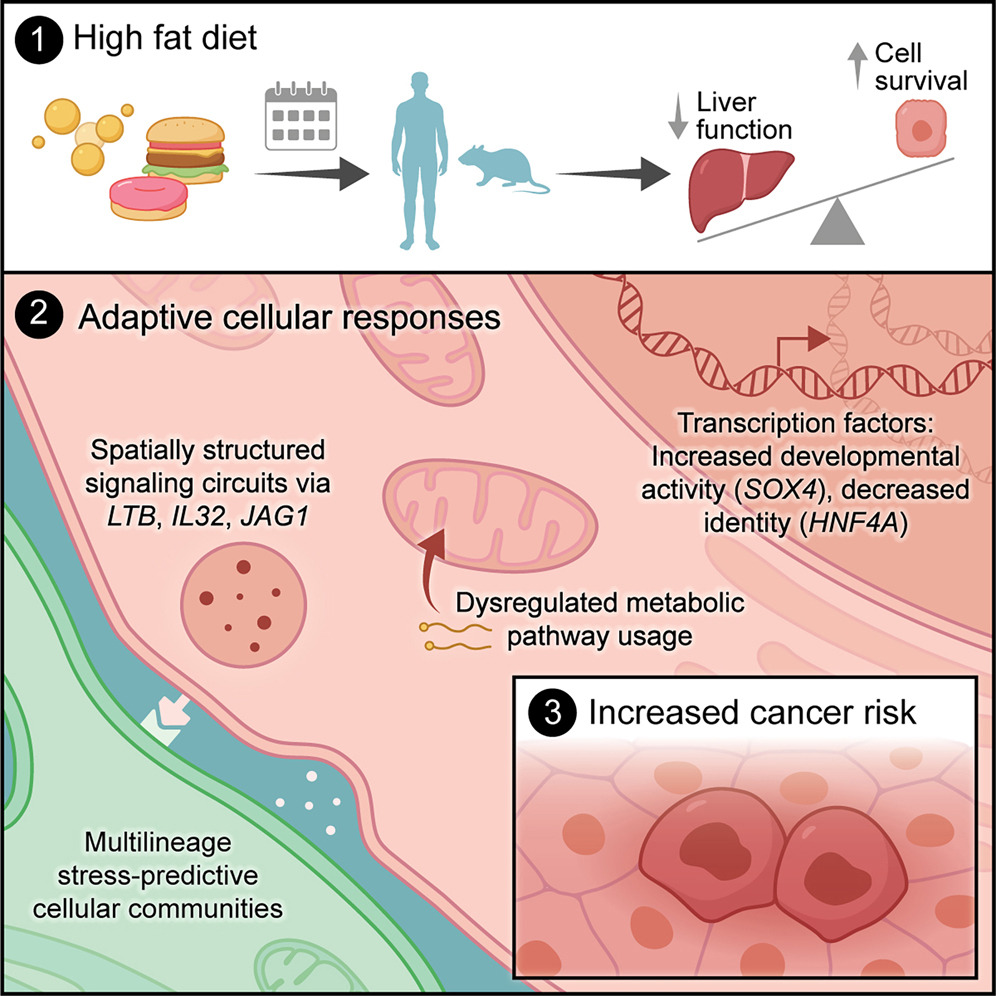
在慢性应激状态下,细胞必须缓冲环境干扰以维持自身存活和组织功能。肝细胞功能非常广泛,包括营养代谢、蛋白质分泌和化学解毒。此外,它还具有强大的再生能力,如在急性损伤(如三分之二肝脏切除)后能恢复正常的肝脏质量和功能。然而,慢性应激会导致这种内在的再生能力缺陷,导致组织损伤。例如,长期摄入致肥胖饮食会诱发代谢功能障碍相关的脂肪性肝病(MASLD,旧称NAFLD/NASH),这影响了全球超过33%的人。MASLD能够进一步发展为代谢功能障碍相关脂肪性肝炎(MASH),其表现为肝脏炎症、纤维化、肝硬化、器官衰竭及肝细胞癌(HCC),其中HCC是癌症减寿的第二大病因(小编注:HCC发病年龄相对较早(常见于40-70岁),且预后较差,很多患者在壮年或中年时期去世,导致每个人损失的寿命年数更多)。
流行病学研究表明,MASH纤维化程度的增加预示着HCC风险的增加。然而,癌前阶段的体细胞突变特征仅在肝硬化患者而非早期的纤维化阶段患者中出现。同样,在以MASH为主的队列中,体细胞突变主要富集在代谢酶相关基因上,而非HCC的关键驱动基因。有趣的是,在其他器官的研究中发现,环境应激能够通过“非突变”方式驱动长期的功能障碍和肿瘤发生,如胰腺和皮肤上皮组织在应对炎症、以及肠道干细胞在应对高脂肪饮食(HFD)时,在转录和表观遗传水平上发生适应性改变(小编注:皮肤和胰腺的上皮干细胞具有“炎症记忆” 。当这些组织经历急性炎症后,即使炎症消退,上皮干细胞仍会在表观遗传水平上维持关键应激反应基因的染色质可及性。在皮肤中,这种开放的染色质状态使得细胞在遇到二次刺激(如伤口或促癌刺激)时,能更迅速地转录应激基因以加速伤口愈合,但这种高度敏感的状态也显著增加了癌症风险。在胰腺中,当遭遇二次炎症时,胰腺细胞能迅速启动保护性程序(ADM),限制损伤、促进修复。但这种保护性适应同时也创造了促瘤微环境,与致癌突变协同,极大加速肿瘤发生。高脂饮食(HFD)诱导肠道干细胞(ISCs)及其子代中PPAR-δ程序的大量表达,这种非突变性的改变赋予了非干细胞原本不具备的“类干细胞”自我更新能力(干性)。此外,HFD驱动CPT1A等酶介导的脂肪酸氧化。这种代谢模式的改变不仅增加了ISCs的数量,还使得这些处于应激状态的细胞在丢失抑癌基因时,形成肿瘤的能力显著增强。参考文献:[1] Del Poggetto E, Ho IL, Balestrieri C, et al. Epithelial memory of inflammation limits tissue damage while promoting pancreatic tumorigenesis.Science. 2021.[2] Beyaz S, Mana MD, Roper J, et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors.Nature. 2016.[3] Naik S, Fuchs E. Inflammatory memory and tissue adaptation in sickness and in health.Nature. 2022.[4] Naik S, Larsen SB, Gomez NC, et al. Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature. 2017.)。据此,研究人员假设肝细胞在应对慢性应激时,也可能产生一种逐渐失调的细胞状态,这种状态虽然并非完全由遗传基因所决定,却为肿瘤发生奠定了基础。
过去针对MASLD的研究多聚焦于特定基因敲除或免疫亚群清除下的组织学与器官生理功能研究、细胞死亡的诱因(如活性氧、未折叠蛋白反应或脂毒性)等领域。相比之下,研究人员对于在慢性应激下存活细胞的表型变化及其动态演变过程仍知之甚少,如:(1)当细胞暴露于慢性应激时,哪些通路被激活,细胞内环境如何平衡?(2)早期应激反应是如何导致了肿瘤发生等后果?(3)哪些通路介导了细胞应对应激的适应性反应?
近期,一篇发表在Cell上的题目为“Hepatic adaptation to chronic metabolic stress primes tumorigenesis”的文章证明了,肝细胞对慢性代谢应激的反应会通过干扰细胞的功能和稳态,从而为肿瘤发生奠定基础。研究人员对饮食诱导肝脏损伤的HFD小鼠模型进行了纵向单细胞多组学分析,以研究从早期肝脂肪变性到自发性肿瘤发生的整个病理过程。基于这些数据,并结合对人类MASLD/HCC队列的分析,研究人员定义了肝细胞活性的渐进式再平衡现象,具体表现为早期发育和与细胞存活相关基因的上调,同时伴随着细胞标志性功能的衰退,如谱系决定性转录因子(TFs)、限速酶以及免疫调节分泌蛋白的表达降低。此外,研究人员还验证了在慢性应激中驱动功能紊乱、增强肝细胞的增殖能力从而驱动肿瘤发生的调控因子。此外,通过分析人类肝组织微阵列的空间转录组数据,研究人员发现细胞外在的、空间结构化的信号互作在肝细胞应激反应中具有重要作用。总之,这篇文章揭示了细胞应对慢性应激的纵向响应机制,并将其与癌症发生和预后建立联系。

敲黑板啦!
1.HFD诱导肝细胞激活应激反应与再生程序,并促进肿瘤发生
2.酮体生成限速酶HMGCS2的下调是促进肿瘤发生的关键代谢驱动因素
3.SOX4与RELB为应激反应的核心转录因子
4.慢性应激通过表观遗传与多细胞信号枢纽促进肝癌发生
研究结果
1、代谢应激重塑了肝细胞的促生存和核心功能定位点
研究人员构建了高脂饮食(HFD)诱导的肝损伤小鼠模型(慢性应激的典型模型)。HFD饮食的C57BL/6小鼠表现出了与MASH患者相似的全身性、组织学和功能性并发症,包括脂肪变性、炎症、纤维化以及自发性肝细胞癌(HCC)的发生(图1A-1D和图S1)(小编注:细胞角蛋白8/18(cytokeratin8/18,CK8/18)是肝细胞骨架的关键蛋白,也是识别肝细胞气球样变的指标,发生气球样变的肝细胞CK8/18的表达呈阴性,而正常肝细胞则呈阳性,肝细胞气球样变是NASH的重要病理特征(参考文献:Tzouanas CN, Shay JES, Sherman MS, et al. Hepatic adaptation to chronic metabolic stress primes tumorigenesis. Cell.2026;189(2):435-460.e28.)。此外,该HFD小鼠形成了自发性肝肿瘤,并且其组织学和转录组特征与HCC相似,包括HCC诊断性生物标志物和的肝癌标志物(图S1)。为了探究细胞对慢性代谢应激的动态反应,研究人员对HFD组和对照饮食(CD)组小鼠进行了纵向单细胞转录组及表观遗传组学分析,涵盖了所有主要的实质、基质和免疫亚群。
研究人员首先聚焦于MASLD中肝细胞的渐进性应激反应(小编注:渐进性应激反应通常指机体在持续或反复的应激源作用下,逐渐出现的一系列生理、心理和行为变化的综合反应),根据基因表达的时间轨迹,定义了四种表达程序:(1)“纵向增加”和(2)“持续上调”分别描述随着慢性应激的暴露,基因表达逐渐增加,以及随时间推移持续上调;(3)“纵向减少”与(4)“持续下调”分别描述慢性应激下基因表达逐渐降低,以及随时间推移持续性下调。
通过对纵向增加与持续上调程序的分析,研究人员发现长期代谢应激会诱导维持细胞存活及抑制凋亡的相关基因(如抗细胞死亡基因Bcl2l1、Cd1d1和Cd59a;再生相关基因Cdkn1a和Tcf4)的表达增加。特定的胆固醇合成酶(如Hmgcr、Hmgcs1)及β-氧化酶(如Cpt1a)基因的表达水平也显著上调(图1E、1F、1I和1J)。基因集富集分析(GSEA)结果表明,与细胞周期调控、凋亡信号负调控、WNT信号通路及β-氧化相关通路被显著富集(图1G和1K)。值得注意的是,尽管这些表达程序最初源于非癌变肝细胞对慢性应激的适应性反应,但在饮食诱导的自发性肝肿瘤中,这些基因的表达水平也显著增加(图1H和1L)。相比之下,“纵向减少”与“持续下调”程序包含维系肝细胞功能特征及稳态的核心基因,包括主谱系调控因子Hnf4a、多种代谢酶(如生酮作用限速酶Hmgcs2;尿素循环限速酶Cps1)、分泌蛋白(如补体和凝血因子)以及肿瘤抑制因子(图1M-1T)(小编注:补体是一种血清蛋白质,活化后具有酶活性、可介导免疫应答和炎症反应。可被抗原-抗体复合物或微生物所激活,导致病原微生物裂解或被吞噬。)。其中,酮体生成关键酶Hmgcs2表达的显著下调尤为关键:(1)生酮作用位于脂质氧化的下游;(2)生酮作用与胆固醇合成均需要利用脂质氧化的副产物——乙酰辅酶A(CoA);(3)许多胆固醇合成酶(包括限速酶Hmgcr)随慢性代谢应激而增加。这提示了,肝细胞在慢性应激下,可能优先进行有利于细胞自身生存和增殖的路径(如胆固醇合成)。与此一致的是,这些基因在饮食诱导的自发性肝肿瘤中也表现出显著下调(图1P和1T)(小编注:这里指的就是“纵向减少”与“持续下调”基因,作者是想说从早期脂肪变性到自发肝癌发展过程中,慢性代谢压力会激活肝细胞的两类(上调或下调)核心程序。上一段是在描述上调的程序基因,这一段是在描述下调的基因。)(小编注:胆固醇合成对于细胞自身生存和增殖的意义在于:①胆固醇是真核细胞膜的基本组分,对于处于慢性应激或快速增殖状态的肝细胞,自给自足地合成胆固醇能确保在不依赖外界脂质供应的情况下,拥有源源不断的原料来构建子代细胞的膜系统;②胆固醇在细胞膜上会聚集形成高度有序的微结构——脂质筏,脂质筏是多种致癌信号通路(如WNT通路、Hedgehog以及EGFR等)的组装中心,高水平的胆固醇合成有助于在胞膜上搭建更多的“接收塔”,使细胞对环境中的生长因子和存活信号更加敏感,从而维持其在恶劣环境下的持续增殖;③胆固醇合成途径的中间产物(如类异戊二烯)是Ras和Rho家族蛋白发生异戊二烯化修饰所必需的。这些蛋白的激活是驱动细胞周期循环和骨架重塑的关键分子引擎。正常情况下,肝脏氧化脂肪酸产生酮体供应大脑等器官(利他),但在慢性应激下,肝细胞通过强化胆固醇合成,将代谢资源从“支持全身生理”转化为“构建自身膜结构、搭建促癌信号平台并激活增殖蛋白”,从而完成了从功能性肝细胞向自私增殖细胞的转型。)。
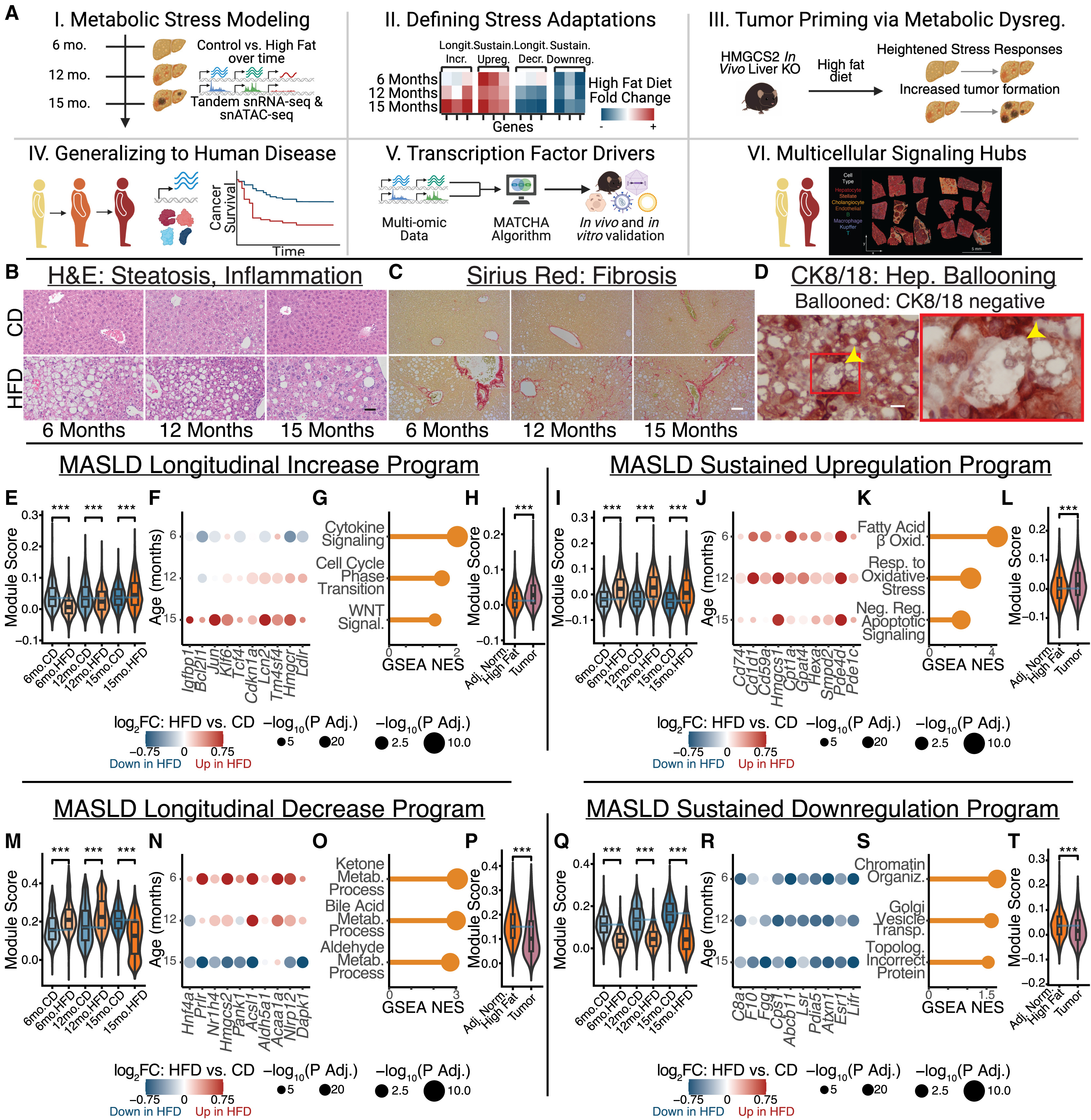
图1. 慢性代谢应激肝细胞的动态反应

图S1. 代谢应激诱导的小鼠功能、器官级和组织级表型特征(对应图1)
2、肝细胞纵向应激反应和功能障碍的多模态验证
与转录组学变化一致,肝细胞“纵向减少”与“持续下调”基因的蛋白质水平丰度降低,具体包括:(1)主谱系决定转录因子HNF4A(小编注:HNF4A是肝细胞中最为核心的转录因子,被誉为肝脏身份的“总开关”。它通过结合基因组的特定区域,驱动肝脏特异性功能基因(如解毒、凝血、物质代谢)的表达。在慢性代谢压力下,HNF4A蛋白丰度的降低意味着肝细胞发生了“去分化”,这不仅导致肝脏稳态功能的丧失,更使肝细胞摆脱了成熟态的生长抑制,从而获得类似祖细胞甚至肿瘤细胞的增殖潜力,是肝癌前病变的核心标志之一);(2)其他功能标志物如EGFR和A1CF(小编注:EGFR:作为细胞表面的受体与表皮生长因子(EGF)等配体结合,刺激细胞生长、分裂和分化;A1CF:作为APOBEC1酶的辅助因子,协助其在mRNA中进行脱氨作用);(3)由肝细胞产生的各种分泌蛋白(包括临床生物标志物ALB)均呈现下降趋势(图2A-2J和图S2A)(小编注:b-catenin在图2B中主要是作为细胞膜的maker,用来界定细胞边界,其亚细胞定位主要在细胞膜;此外,β-catenin是经典WNT/β-catenin信号通路的核心效应分子,在正常状态下,β-catenin主要作为E-cadherin/catenin复合物的一部分,定位于细胞膜,当Wnt信号通路激活时,β-catenin得以稳定并入核,启动下游靶基因转录,详情如下:

在缺乏Wnt信号的情况下,细胞质中的β-catenin被Axin、APC、丝/苏氨酸激酶GSK-3和CK1、蛋白磷酸酶2A(PP2A)以及E3泛素连接酶β-TrCP组成的蛋白复合物经磷酸化和泛素化修饰后被降解。在细胞核中,T细胞因子(TCF)因与抑制因子Groucho结合而处于非活性状态。当Wnt信号通路激活时,Wnt与其受体的结合诱导Axin与磷酸化脂蛋白受体相关蛋白(LRP)结合。进而使复合物降解,促使β-catenin在胞质的积累和稳定,解除磷酸化的β-catenin易位到细胞核,与TCF结合形成复合物,从而上调下游靶基因的表达。参考文献:Nusse R, Clevers H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell.2017;169(6):985-999.)。与此同时,作者测到WNT靶基因GLUL的表达增加(小编注:GLUL表达增加说明肝细胞身份发生了重编程。GLUL表达增加与HNF4A的减少共同表明:在慢性压力下,肝细胞一方面关闭了维持其成熟身份和正常功能的核心程序(由HNF4A主导),另一方面激活了驱动其适应、增殖并最终可能导致癌变的通路(由WNT/β-catenin主导))(图2C-2E)。在体外利用高脂培养基培养人肝癌细胞系HepG2时,研究人员观察到了转录水平的变化、脂质积累以及蛋白质合成的变化,这些结果均与其在体内的研究一致(图S2B-S2H)。
此外,鸟枪法脂质组学分析(小编注:鸟枪法脂质组学是一种基于质谱的高通量脂质分析技术。不同于传统的脂质检测,鸟枪法通过直接进样的方式,在分子层面扫描并定量数以千计的脂质物种。它能精确揭示脂质分子的微细结构变化,例如甘油三酯碳链的延伸或不饱和度的改变)显示了肝脏脂质发生广泛重塑。在HFD饮食6个月和12个月时,甘油三酯水平显著升高,这与组织学定量结果相吻合(图2K和S1B)。甘油三酯总碳链长度的逐渐增加不仅与延伸酶Hacd1的上调一致(图S2I和S2J),也符合肝再生过程中碳链长度增加的规律。
拓展阅读
肝再生过程中碳链长度增加的规律
肝再生过程中碳链长度增加的规律表现为:从早期(12-24h)以20碳以下的长链脂肪酸能量储备为主,逐渐过渡到增殖期(48h)以20碳以上的超长链脂肪酸结构重塑为主。这一过程由包括Hacd1在内的延伸酶系精密调控,确保了肝脏在修复过程中能量供应与物质构建的完美衔接。
1.肝再生过程中的碳链长度演变规律
肝脏在再生过程中,其甘油三酯的脂肪酸组成会经历从“长链”向“超长链”的转变:
(1)再生早期(TRAS阶段,12-24小时):肝脏处于再生相关性脂肪变性(TRAS)时期,此时积累的甘油三酯主要由长链脂肪酸(Long-chainfattyacids,LCFA)组成。这些脂肪酸的碳原子数通常少于20个(主要归类为C1组脂肪酸)。
(2)细胞增殖期(48小时):随着再生进入肝细胞大量增殖阶段,甘油三酯的成分发生显著迁移,其脂肪酸组成从长链转向了超长链脂肪酸(Ultra-long-chainfattyacids,VLCFA)。此时上调的脂肪酸(C2组)碳链长度普遍超过20个碳原子。
2.延伸酶Hacd1与碳链增长的分子关联
Hacd1(3-hydroxyacyl-CoAdehydratase1)是脂肪酸延长循环中的关键限速酶之一。甘油三酯总碳链长度的逐渐增加与Hacd1基因表达的上调在时间轴上是高度一致的。Hacd1的上调直接驱动了脂肪酸链的延伸过程,使得肝脏能够合成更多碳链更长、不饱和度可能更高的脂肪酸,以满足增殖期细胞膜构建和信号传导的需求。
3.这种碳链长度的增加并非随机,而是为了适应不同阶段的代谢需求
(1)早期(C<20):较短的长链脂肪酸(如C16,C18)通常包含饱和成分,结构更稳定,主要作为能量库,通过β-氧化为即将开始的细胞分裂提供充足的ATP。
(2)后期(C>20):增殖期增加的超长链脂肪酸多为不饱和脂肪酸。这些超长链成分对于构建高度流动性的细胞膜磷脂至关重要,同时也可能作为再生调节信号分子的前体。
参考文献:
[1] Li YN, Sun FF, Ouyang F, et al. Alterations in liver triglyceride profiles in CCl4-induced liver regeneration.Biochem Biophys Res Commun. 2024
在饮食诱导15个月时,与对照相比,HFD饮食组的胆固醇及鞘脂类脂质丰度增加最多(图2L)。肝脏中胆固醇及胆固醇相关脂质丰度的增加,与胆固醇合成限速酶Hmgcr(胆固醇合成限速酶)、Hmgcs1及Ldlr的上调,以及生酮作用限速酶Hmgcs2(生酮限速酶)的下调相一致(图1F、1J和1N)。HFD组血浆胆固醇含量的增加也进一步佐证了上述结果(图S2K)。此外,鞘脂相关酶Smpd2和Hexa的上调进一步揭示了肝脏的转录层面与代谢表型的关联:鞘脂介导代谢相关脂肪性肝炎(MASH)的进展和肿瘤的免疫逃逸。
拓展阅读
鞘脂介导代谢相关脂肪性肝炎(MASH)的进展和肿瘤的免疫逃逸
(1)代谢紊乱相关性脂肪性肝炎(MASH)导致细胞应激、肝损伤、炎症和纤维化。鞘脂在膜结构和细胞过程中发挥重要的作用。肝脏作为鞘脂合成的主要场所,极易受到鞘脂毒性的影响。鞘脂代谢失调是MASH的关键驱动因素。神经酰胺是鞘脂代谢的中心,主要通过两种途径产生:从头合成和鞘磷脂水解。值得注意的是,MASH患者的神经酰胺水平升高,鞘磷脂无显著变化,而此前对鞘磷脂水解在MASH进展中的重要作用尚不清楚。研究发现MASH患者的肝脏脂毒性会触发依赖sirtuin1(SIRT1)蛋白的鞘磷酸二酯酶3(SMPD3)上调,促进SMPD3在质膜上催化鞘磷脂水解从而升高神经酰胺水平,这破坏了膜鞘磷脂-神经酰胺的平衡,进而促进了胞膜依赖的脂质摄取和促炎促纤维化细胞外囊泡(EVs)的分泌,促进疾病进展,从而加剧炎症和纤维化。

(2)KRAS基因突变驱动的癌细胞,能够塑造抑制性肿瘤微环境,从而得以生存和生长。脂质对维持膜稳态、信号传导和能量产生至关重要。快速增殖的癌细胞需要持续的脂质供应以支持其生长及适应微环境。参与脂质合成与摄取的代谢基因是致癌性改变的下游效应因子,通常在肿瘤中表达上调。脂质代谢的这些改变也受肿瘤微环境和机体整体代谢的影响。缺氧的肿瘤细胞会增加脂质摄取以补偿脂肪酸去饱和作用的减弱;肿瘤微环境中的饱和脂质能够保护癌细胞免受氧化损伤。此外,在高脂饮食的小鼠模型中,从食物中获取的脂质会促进肿瘤发生、转移和免疫逃避。在某些情况下,脂质的促肿瘤效应是通过改变肿瘤微环境中免疫细胞功能介导的。但此前尚未确定促进肿瘤发生和免疫逃避的特定脂质种类。研究发现鞘脂的从头合成是癌症免疫逃逸的关键通路。阻断癌细胞中的糖鞘脂合成的关键酶UGCG依赖的鞘脂合成致使糖鞘脂耗竭,能够增强肿瘤微环境中自然杀伤细胞和CD8+T细胞的抗肿瘤增殖能力,产生更多的干扰素-γ(IFN-γ),同时癌细胞中糖鞘脂的耗竭会导致其细胞膜表面的IFN-γ受体1(IFNGR1)水平显著上升,增强癌细胞对免疫细胞释放的IFN-γ信号的敏感性;进而导致其下游信号通路JAK-STAT1的过度激活,最终导致IFN-γ诱导的癌细胞生长停滞和促炎反应,从而抑制肿瘤生长。
肿瘤发生前鞘脂的增加表明,肝脏在肿瘤发生前就已发生代谢重编程,这种代谢转变可能与未癌变肝细胞中参与免疫逃逸的转录(如Cd59a和Cd1d1)相协同(小编注:Cd59a和Cd1d1这两个基因本身不直接参与鞘脂合成,他们是肝细胞在慢性代谢应激下激活的免疫逃逸相关基因(图1J)。Cd59a主要负责保护细胞免受补体攻击,从而保护细胞不被补体系统溶解。Cd1d1则通过呈递脂质抗原激活自然杀伤T细胞(NKT),参与免疫调节和病原体清除。原文这段话的意思是:在肿瘤发生前,高脂饮食小鼠的肝脏中就发生了鞘脂相关酶Smpd2和Hexa上调(图1J),导致了鞘脂在肝脏中累积(图2L)。引用的参考文献中又提到:(1)鞘脂相关酶Smpd3的上调会促进MASH的进展(Jiang J, Gao Y, Wang J, et al. Hepatic sphingomyelin phosphodiesterase 3 promotes steatohepatitis by disrupting membrane sphingolipid metabolism. Cell Metab. 2025;37(5):1119-1136.e13.),而(2)新生鞘脂的合成增加能保护癌细胞免受NK和CD8+T细胞的免疫监视(Soula M, Unlu G, Welch R, et al. Glycosphingolipid synthesis mediates immune evasion in KRAS-driven cancer. Nature. 2024;633(8029):451-458.)。并且免疫调节/逃逸相关基因Cd59a和Cd1d1发生上调(图1J)。二者具有协同作用,介导非癌变肝细胞的免疫逃逸)。
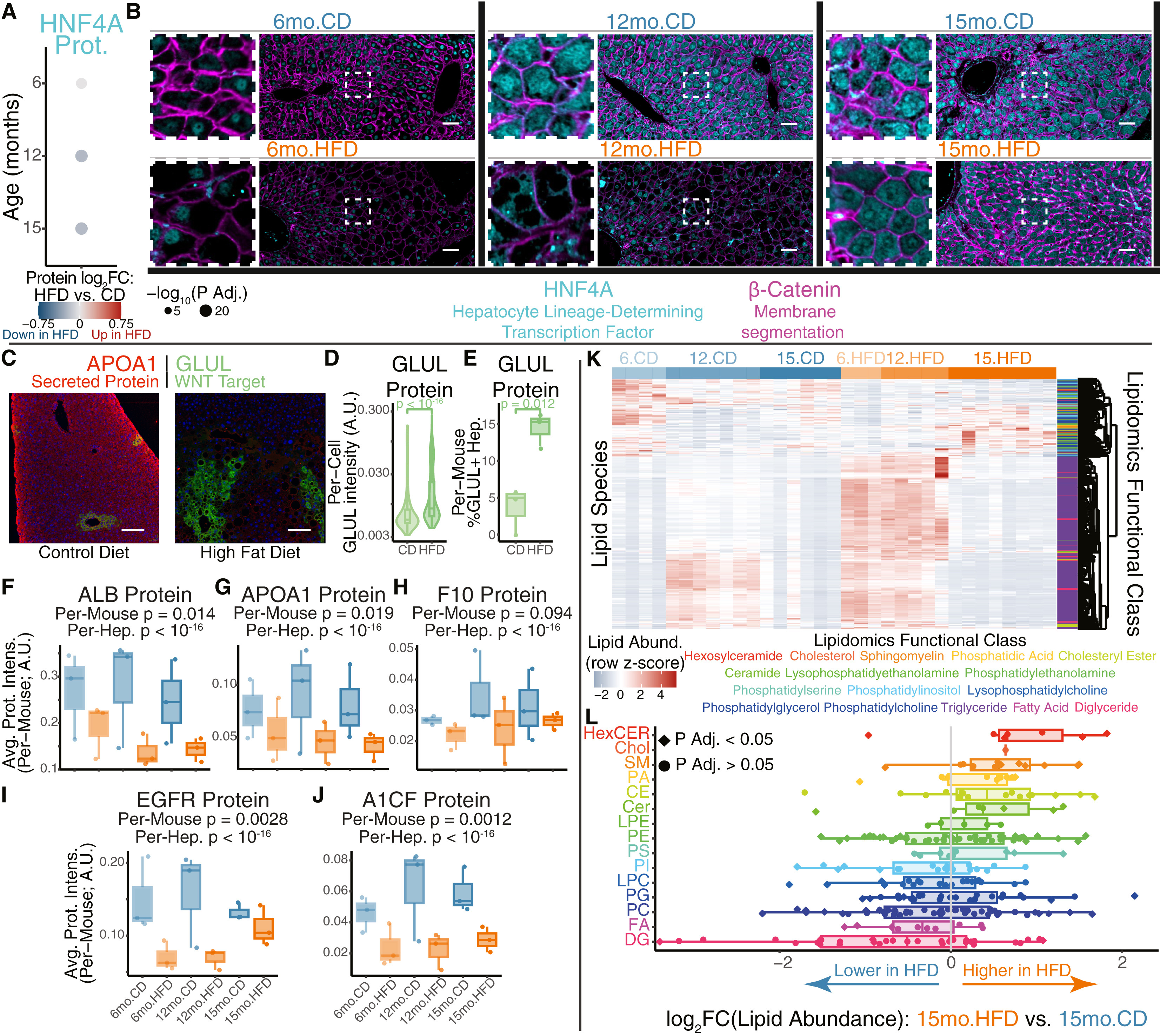
图2. 肝细胞代谢应激反应的多模态验证
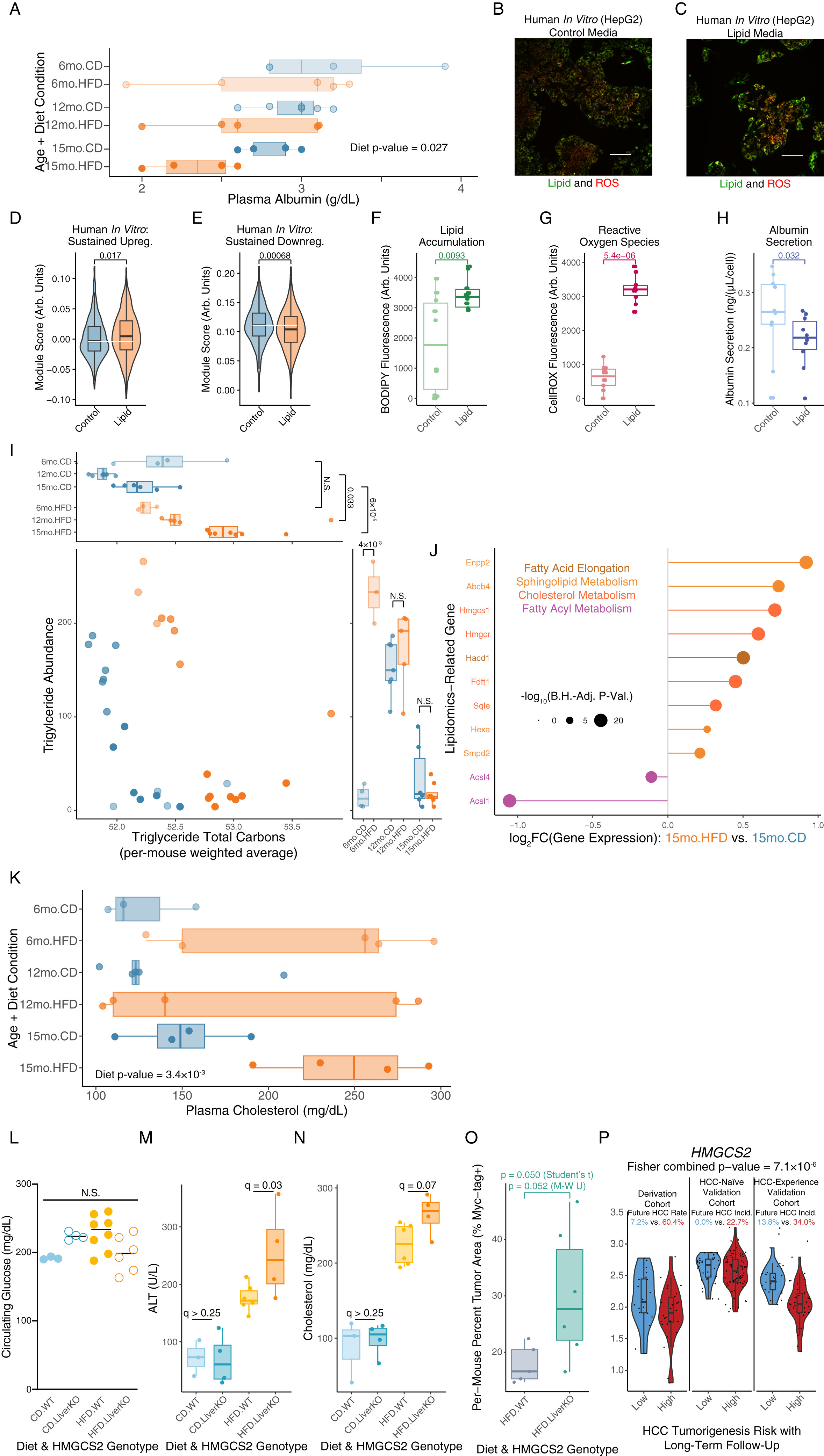
图S2. 人类体外脂质培养、小鼠体内大体脂质组学及小鼠体内肝脏HMGCS2基因敲除实验共同支持肝细胞的应激适应机制(对应图2和图3)
3、代谢途径的失衡加剧应激反应促进肝肿瘤的发生
为了研究代谢应激带来的直接后果,研究人员进一步探讨了饮食驱动的代谢改变是否足以加剧肝细胞的应激反应并恶化肿瘤结局,重点研究了“纵向减少”程序中的关键成员——HMGCS2(3-羟基-3-甲基戊二酰辅酶A合酶2),它是生酮作用的限速酶。在代谢应激驱动下,HMGCS2在转录和蛋白质水平上均表现出显著下降,且较低的HMGCS2表达水平预示着人类HCC患者更差的生存预后(图1N和图3A-3E),此外,已知在人类脂肪性肝炎中,利用脂质氧化副产物产生酮体的过程也会受损。
为了模拟代谢应激驱动的Hmgcs2表达下调,研究人员构建了肝脏特异性敲除小鼠模型(Hmgcs2fl/fl;Alb-Cre)。与预期一致,在HFD的诱导下,所有基因型小鼠均发生脂肪变性,但其体重增长和循环葡萄糖水平与是否敲除Hmgcs2无关(图3F、3G和S2L)。然而,在HFD与HMGCS2肝脏特异性敲除的共同作用下,小鼠的肝损伤程度和循环胆固醇水平显著增加(图S2M和S2N)。HMGCS2免疫组化和空腹循环酮体的减少分别从蛋白和功能层面验证了敲除的有效性(图3H和3I)。
随后,为了探究HMGCS2的缺失是否足以加剧和加速肝细胞的渐进性应激反应,研究人员对不同Hmgcs2基因型和饮食条件下的样本进行了单细胞核RNA测序(snRNA-seq:n=8mice,27119cells)。结果发现HMGCS2的缺失加速了肝细胞的应激反应。具体表现为,在对照组(HFD WT)中,有52%的肝细胞状态映射到经历HFD饮食6个月的应激状态,有44%的肝细胞状态映射到经历了HFD饮食12个月的应激状态。然而,在HFD HMGCS2肝脏特异性敲除组中,映射到HFD12个月的肝细胞(63%)是映射到HFD6个月的肝细胞(30%)的两倍以上(图3J)(小编注:映射是指利用复杂的统计算法,将一个未知状态细胞的全局基因表达谱,与一个预先定义好的“参考细胞图谱”进行比较,从而将该细胞分配或归类到参考图谱中最相似的那个细胞状态或类型中去。此处研究人员首先对“自然应激进展队列”的所有细胞进行单核RNA测序,利用其基因表达数据,绘制出详细的“肝细胞状态地图”。然后,他们将HFD WT和HFD LiverKO小鼠的每一个肝细胞,放到这张地图里去比对。算法会判断每个细胞在基因表达特征上,与地图中哪个自然进展状态最相似)。类似地,相对于WT小鼠,HMGCS2的缺失诱导了肝细胞应激反应程序的极端表达(图3K)。HMGCS2缺失+HFD上调了多种胆固醇合成相关基因(如Hmgcs1,Srebf2和Mvk)(图3L),还诱导了细胞功能障碍,表现为发育相关标志物、肝癌相关细胞间信号传导蛋白的上调,以及肝细胞功能相关标志物的下调。
为了探究HMGCS2的缺失是否会促进肿瘤发生。研究人员通过流体动力学尾静脉注射技术(小编注:流体动力学尾静脉注射技术是一种在活体动物(如小鼠)中实现高效非病毒转基因的物理手段。其核心原理是“体积压力效应”:研究者在短时间内(通常为5-8秒)将大剂量的质粒DNA溶液(约占小鼠体重的8%-10%)快速注入尾静脉。瞬时的高流体压力会导致血液逆流并引发心脏的短暂负荷,进而迫使门静脉产生高压,使肝脏血窦扩张、肝细胞膜产生瞬时微孔,从而将DNA质粒直接推入肝细胞核内)将编码hMET,组成型激活的S45Y-CTNNB1及Sleeping Beauty转座酶的质粒导入HFD小鼠的肝细胞内以诱导HCC的发生(图3M)。结果显示,在慢性代谢应激下,肝脏缺失HMGCS2直接导致了肿瘤发生的增加,表现为单只小鼠肝脏肿瘤面积增大(图3N和3O)以及单个肿瘤体积的增加(图3P)。此外,HFD喂养时间也影响了肿瘤发展的程度:即使在引入癌基因前进行2周的HFD喂养,也增加了敲除组小鼠的肿瘤发生,但程度相对较轻(图S2O)。一致的是,通过对三个人类临床队列的分析发现非癌肝组织中HMGCS2的表达降低,这预示着未来长达15年中患肝癌风险增加(图S2P)。这些结果证明了肝细胞应激反应的累积特性,为未来的肿瘤发生奠定了基础。
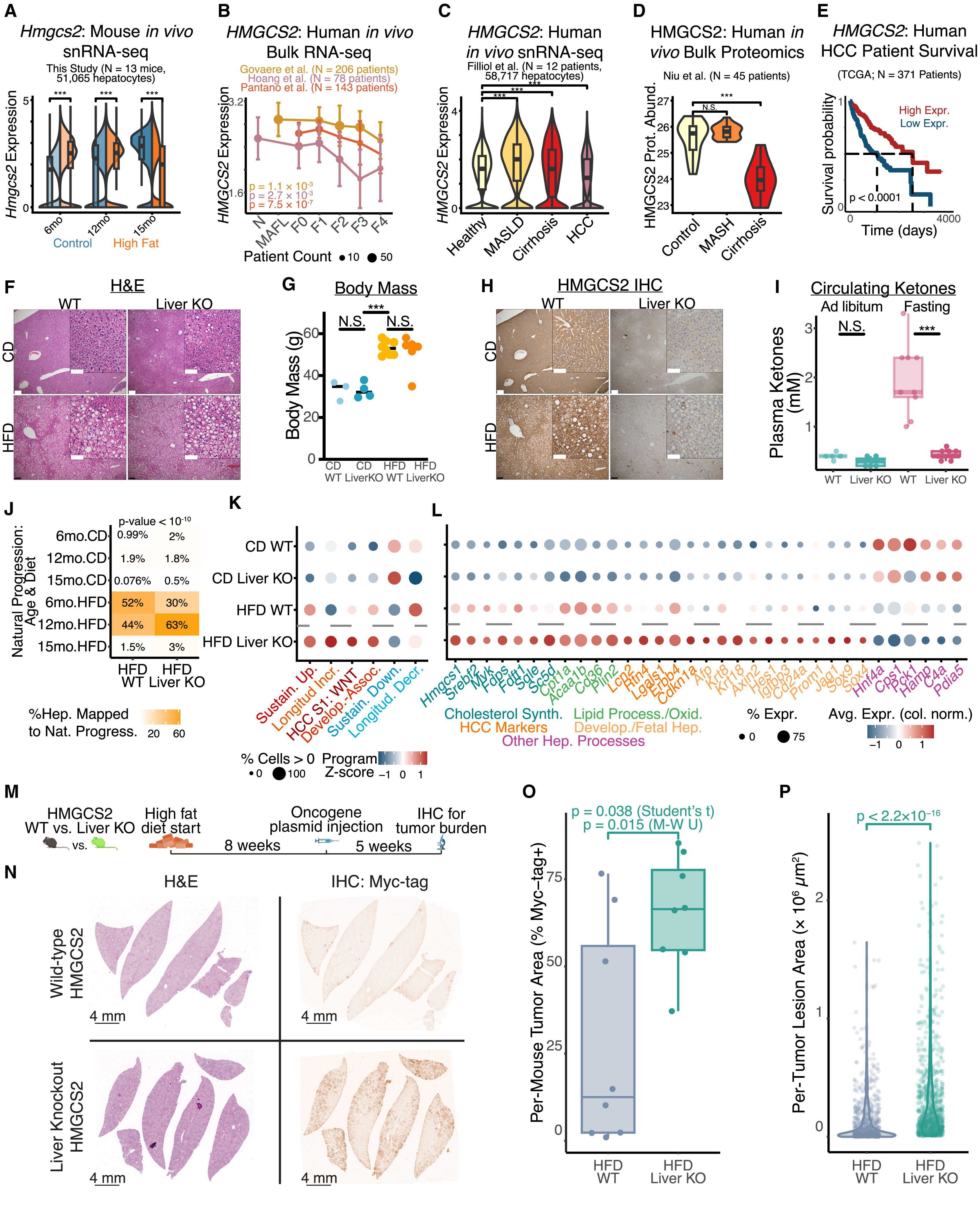
图3. HMGCS2作为肝细胞应激反应及肿瘤启动调节因子的体内验证
4、转化前应激反应延展至人类疾病并可预测癌症生存期
鉴于HFD小鼠模型在生理特征上与人类MASLD/HCC(图S1)高度相似,且代谢重编程对小鼠肝癌具有显著的功能性影响,接下来研究人员探讨了细胞慢性应激反应程序与公开的人类MASLD/HCC数据之间的联系(图4A)。通过对批量RNA-seq和snRNA-seq数据分析发现,随着MASLD严重程度的增加,“纵向增加”程序在不同队列中均显著升高(图4B-4C)。同样,在人类肝组织蛋白质组学研究中,该程序的蛋白水平丰度也随MASLD的病程进展而增加(图4D)。此外,“纵向增加”程序在人类肝癌中进一步富集,并且能对肝癌患者的生存预后进行分层:其高表达预示着更差的临床结局(图4C-4E)。
“持续上调”、“纵向减少”和“持续下调”这三类程序在小鼠和人类中,与疾病严重程度、肿瘤表型及患者生存预后显著相关(图4F-4Q)。正如预期,“纵向增加”和“持续上调”程序的变化趋势,与“纵向减少”和“持续下调”程序截然相反。跨研究、跨检测手段及跨物种的分析显示,这些综合评分背后是由相似的基因驱动的(图S3A-S3F)。即早期代谢应激(比如MASLD早期)时,WNT相关通路就开始变化,最终驱动HCC阶段WNT通路的激活。
鉴于HFD小鼠出现了自发性成瘤现象,研究人员进一步将小鼠的观察结果与人类癌症发病率建立联系。探究了HCC相关特征如何在尚未形成明显肿瘤的早期MASLD中体现。结果发现,即使在MASLD早期阶段,以下特征已在多组学和跨物种数据被激活:(1)具有肝上皮祖细胞特征的肝脏发育特征;(2)HCC人类患者的S1特征:该特征与WNT通路激活和肿瘤早期复发相关(图S3G-S3P)。有趣的是,研究人员也发现这套慢性代谢应激反应程序在不同诱因导致的肝癌中均存在(图S3Q)。此外,这些程序在急性肝损伤再生(如对乙酰氨基酚过量导致的损伤)中也会被激活(图S3R)。这种跨疾病、跨损伤类型的保守性表明,这套程序代表了肝脏应对多种环境压力时的一种普适性应激反应。
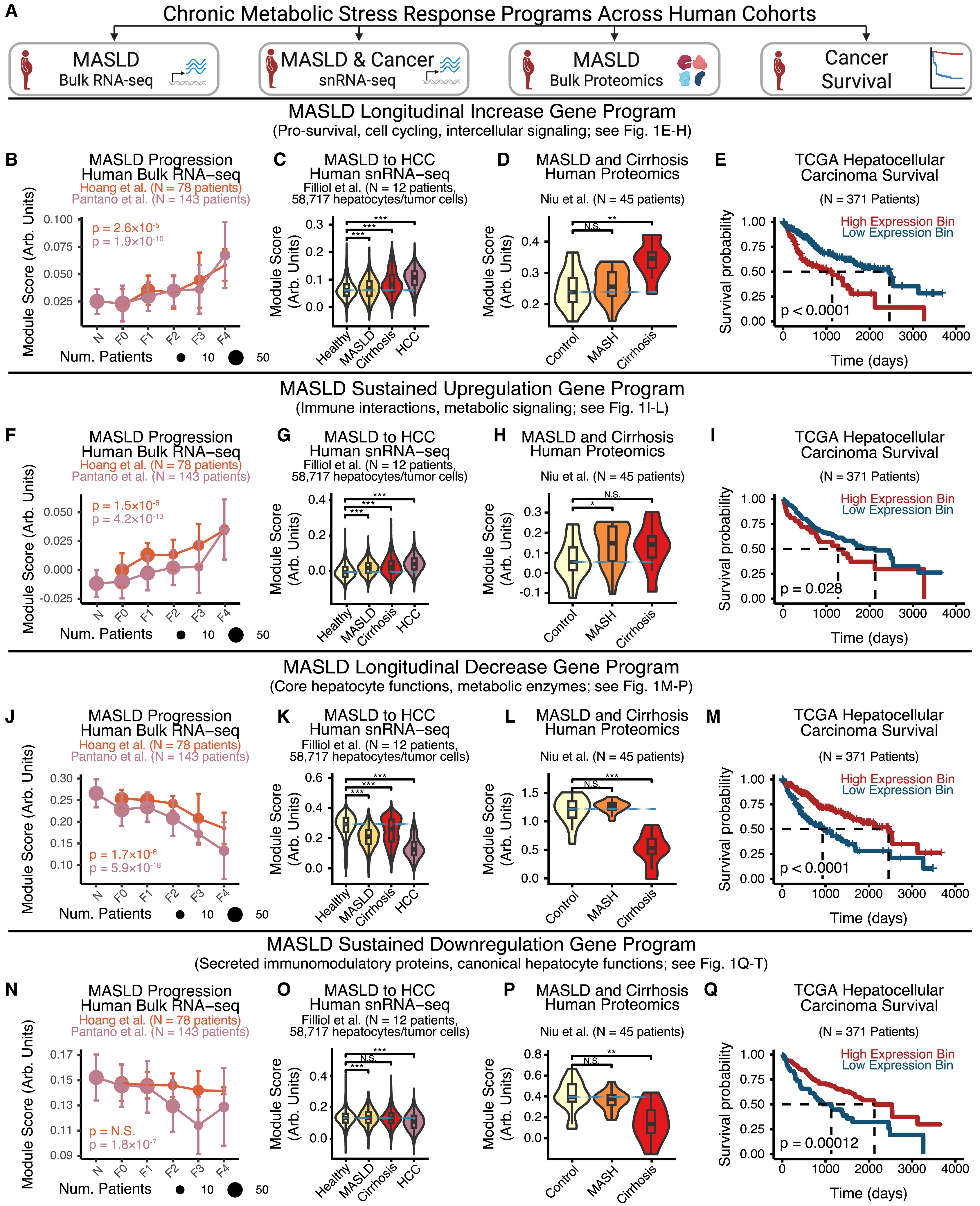
图4. 慢性应激反应延伸至人类队列,并与癌症表型及临床预后密切相关
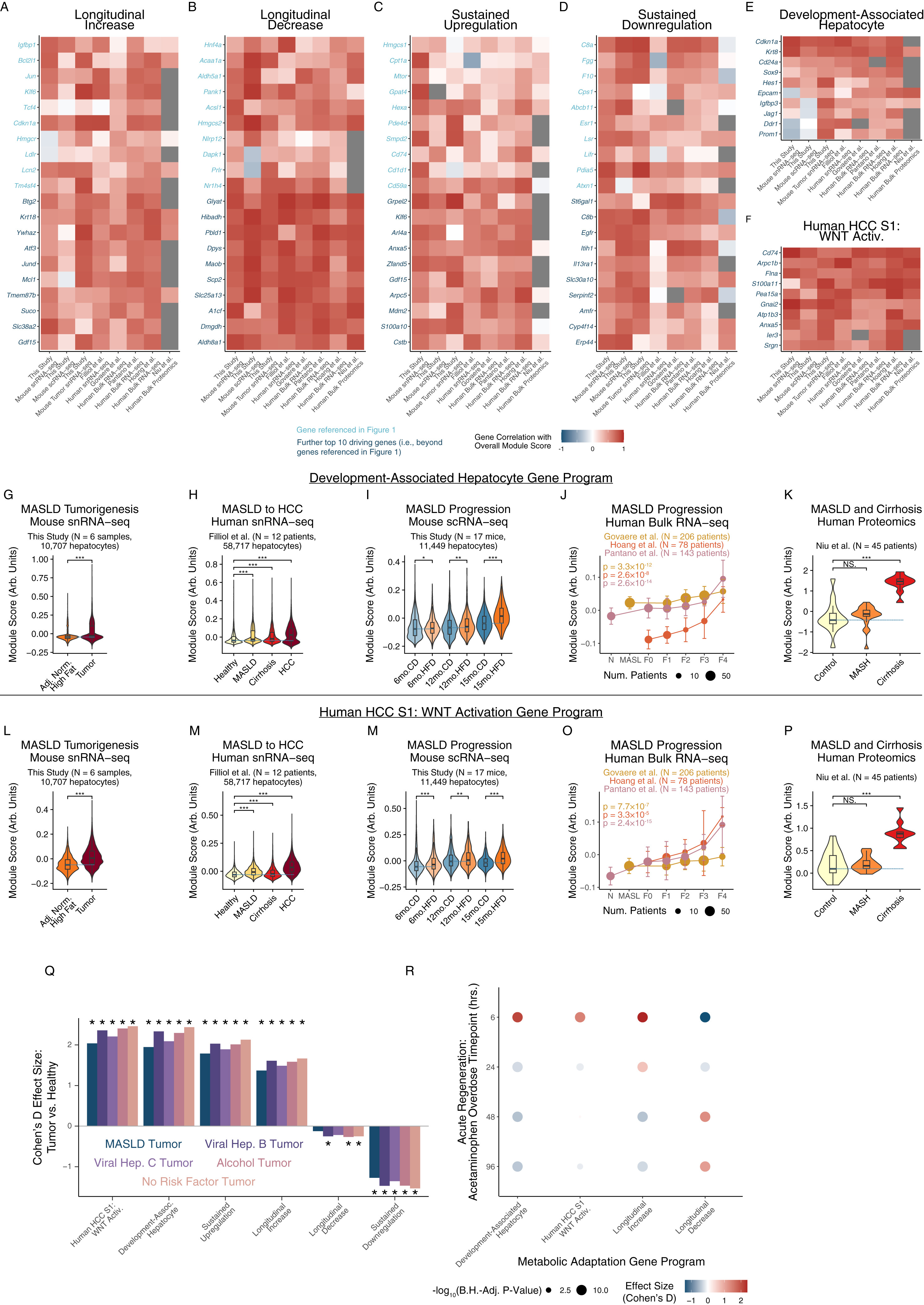
图S3. 代谢应激基因程序的驱动基因(对应图4)
5、慢性代谢应激下的表观遗传失调与WNT通路启动
为进一步鉴定驱动并维持肝细胞应激反应程序的调控机制,通过HFD小鼠的单细胞转座酶可及性染色质测序(snATAC-seq)数据,研究人员发现转录表达与表观遗传的可及性(小编注:表观遗传可及性是指染色质中DNA区域对转录因子、染色质修饰酶和其他调控因子的开放程度。在细胞处于一定状态下时,其基因组DNA上调控元件(如启动子、增强子、沉默子)的染色质的物理构象处于开放状态。这种开放构象允许转录因子、共激活/抑制复合物以及RNA聚合酶等大分子识别并结合其DNA序列,从而启动或调控基因转录)之间具有高度一致性(图S4A)。对全基因组转录因子(TF)结合位点的可及性研究显示,AP-1复合物(小编注:AP-1复合物是一类由Jun和Fos家族蛋白构成的二聚体转录因子。在肝脏病理生理过程中,AP-1是介导“应激驱动型表观遗传重塑”的核心节点。它通过招募染色质重构复合物,在增强子区域诱导持久的开放状态,从而形成“组织炎症记忆”,使肝细胞在脱离原始应激源后仍维持促炎或促癌的转录潜能)位点的可及性随HFD饮食时长的增加而增加,该复合物已被证明在其他器官中介导炎症相关的表观遗传重塑和“组织炎症记忆”(小编注:根据Naik和Fuchs(2022)的研究,在肥胖或高脂饮食诱导的慢性应激下,AP-1复合物(如c-Jun、Fra-1)在脂肪组织中扮演了“先锋转录因子”的关键角色。它能够物理性地开启原本处于关闭状态的促炎增强子,通过募集染色质重塑复合物实现表观遗传重塑;即便在炎症诱因消除后,AP-1仍能像“书签”一样维持这些位点的染色质可及性,从而形成持久的“组织炎症记忆”。这种记忆不仅导致脂肪细胞功能去分化,还通过系统性炎症信号产生跨器官效应(如诱导肝脏病理重塑),使机体在不依赖DNA突变的情况下进入持久的代谢紊乱与致癌预激状态。参考文献:Naik S, Fuchs E. Inflammatory memory and tissue adaptation in sickness and inhealth. Nature.2022,607(7918):249-255.)(图5A)。同样,HFD喂养也增加了参与肝脏再生与发育的TFs(SOX4,SOX9,TEAD,TCF7,LEF1)的基序可及性(小编注:基序可及性是指基因组中特定转录因子结合序列(Motif)所在区域的染色质开放程度。利用snATAC-seq技术检测基序可及性,能够反映细胞在当前表观遗传景观下,对特定调控信号的转录响应阈值。可及性的增加意味着该基因位点已解除核小体限制,处于“预激活”状态)。
研究人员通过伪时间分析(小编注:伪时间分析是基于单细胞组学数据的计算生物学推断方法。通过机器学习算法将离散状态的单细胞表达图谱映射到连续的线性或分支轨迹上,从而模拟生物过程(如细胞分化、恶性转化)随时间轴演进的动态分子特征)进一步探讨了随时间变化的染色质状态(图5B和S4B)。多组学分析再次印证,“纵向减少”程序的基因在HFD喂养早期表现出最大的染色质可及性,而“纵向增加”程序的成员则在末期达到峰值。对基因位点的可及性检测支持了上述伪时间排序结果(图5B-5C),其中,Hmgcs2位点的表观遗传可及性下降支持了先前在慢性代谢应激及HCC中发现的关于该基因在转录、蛋白质及功能水平上失调的结果(图1N、3和图S3)。
为了探究表观遗传的启动,研究人员利用染色质区域间的共可及性分析,来捕获远端增强子与启动子之间的相互作用(小编注:共可及性描述两个或多个基因组区域(如远端增强子与近端启动子)在三维空间中同时处于染色质开放状态的概率或程度。开放染色质区域(如增强子、启动子)在三维空间中可通过染色质环化、拓扑关联结构域(TAD)等机制相互靠近,形成协同调控关系)。以WNT/β-catenin的靶基因Axin2为例,研究人员发现多个远端染色质区域与Axin2的启动子或基因本体具有共可及性,且在HFD喂养过程中,这些区域的可及性均随时间而增加(图5D)。更广泛地说,与WNT信号通路及肝癌相关的基因在HFD喂养早期(6个月)就表现出可及性增加,但此时的转录水平的变化不显著(图5D和5E)。然而,在肿瘤发生后期(15个月),这些基因表达显著上调(图5F)。反之,表现出早期染色质关闭、随后转录下调的基因包括:(1)代谢酶和分泌蛋白;(2)HCC与胎儿肝细胞相关表型的抑制因子;(3)雌激素受体Esr1。早期发现的WNT通路及HCC相关的染色质变化预示着后续转录调控的发生,这表明在代谢应激的早期阶段,可能建立起了一种潜在的、许可性的染色质环境,从而促进后续再生、发育及癌症相关通路的激活(小编注:许可性染色质环境指一种特定的表观遗传稳态。在这种状态下,与再生、发育及癌变相关的关键基因位点处于染色质松弛状态,即便此时由于缺乏足够的激活信号而转录水平较低,但其物理结构的开放为后续信号传导调控基因表达奠定了基础),为后续再生、发育及癌症相关通路的激活奠定了基础。
为了进一步探讨WNT通路失调与HFD诱导的肝细胞应激程序的关系,研究人员汇总了多种WNT干扰模型的数据。结果显示,只激活或抑制WNT通路均不足以驱动完整的肝细胞应激反应程序(图S4C-S4Q)。WNT干扰与应激程序之间的重叠基因主要富集在脂肪酸、胆固醇和胆汁酸代谢通路中(图S4E,S4H,S4K,S4N和S4Q)。同样,在多种模型系统和特征图谱中,仅用脂质处理也不足以增强肝小叶中间区域与WNT相关基因的表达(图S4R和S4S)。这表明,这些应激反应程序可能部分受到WNT信号的影响,但可能受到其他多种分子的共同调控。
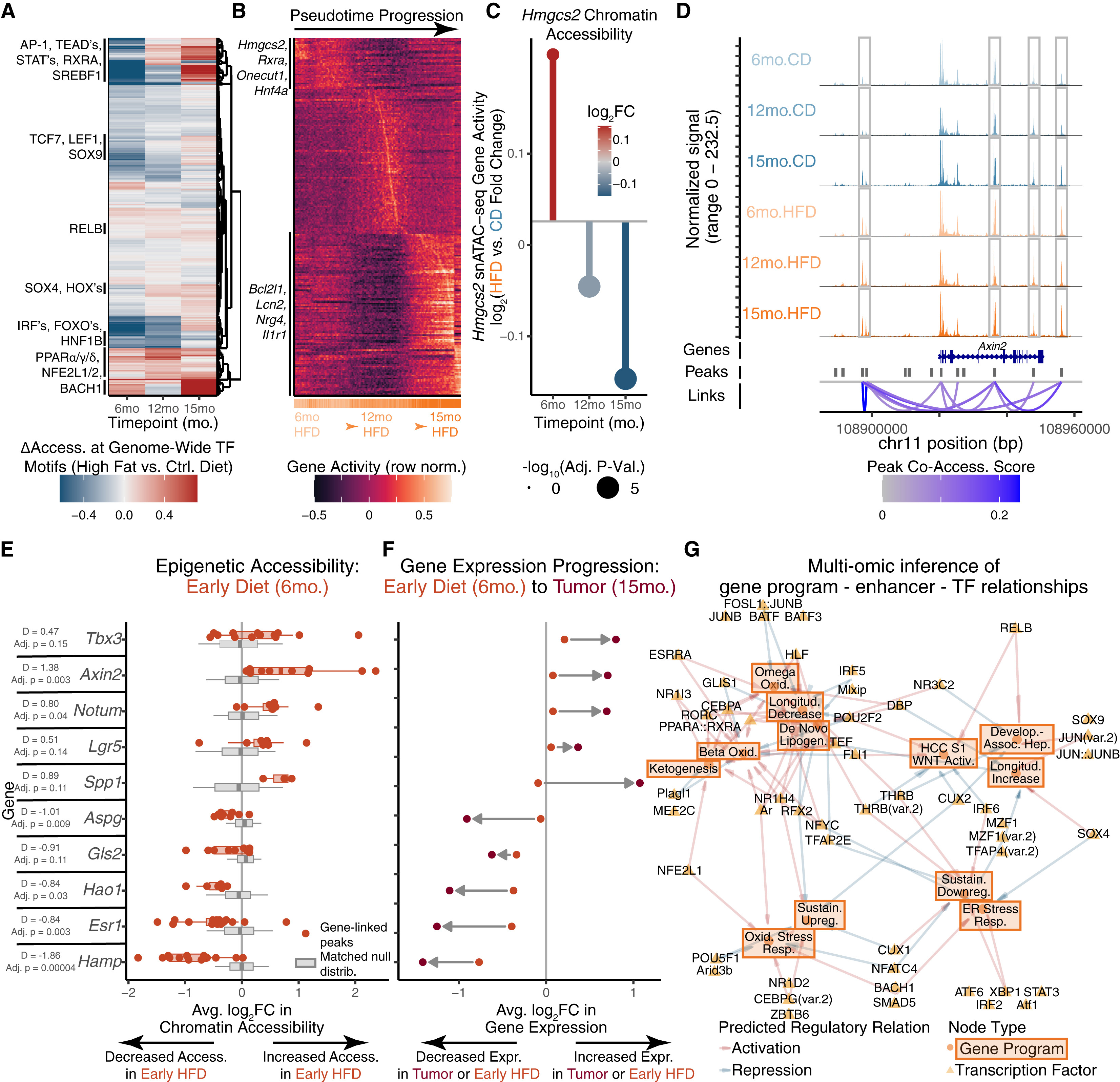
图5. 慢性代谢应激驱动表观遗传轨迹改变及WNT通路启动
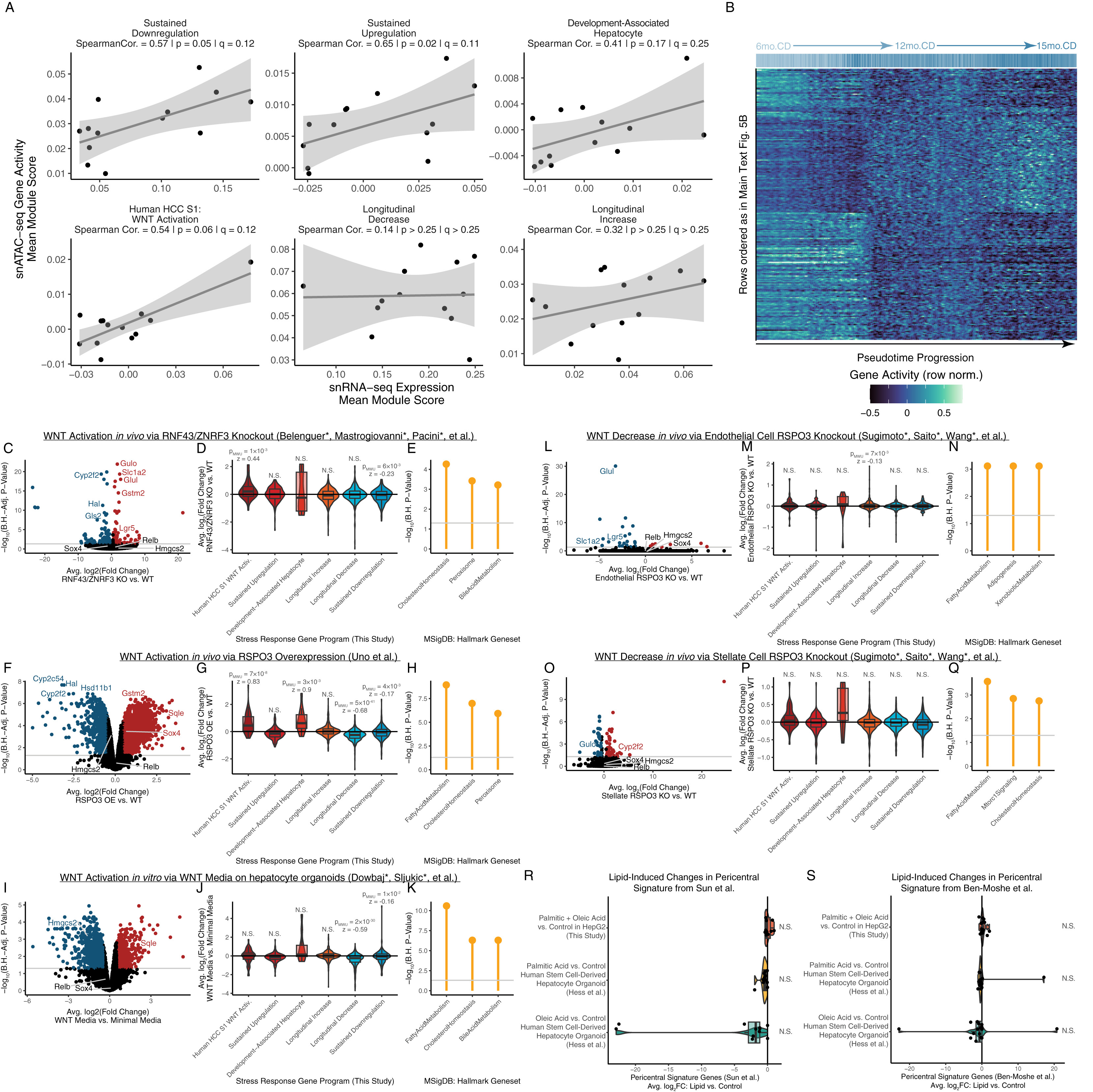
图S4. 肝细胞适应慢性代谢应激的表观遗传轨迹,及其在WNT通路扰动下的关联分析(对应图5)
6、通过MATCHA来预测调控慢性应激反应的TFs
为了探究能够调控肝细胞应激反应的因子,研究人员开发了基于层级关联的多组学转录因果判定工具(Multi-omic Ascertainment of Transcriptional Causality via Hierarchical Association,MATCHA)。该算法利用用户指定的基因程序和多组学数据集(可跨物种),通过对细胞类型及组织特异性基因调控图谱进行相关建模,从而识别不同程序中的核心调控转录因子(TF)。比如,在内质网(ER)应激反应中,MATCHA锁定的排序靠前的TFs是XBP1和ATF6,它们是公认的主调控因子(图S5A-S5G)。β氧化中排名最高的TFs包括FXR/NR1H4和PPARA(两者的激动剂均已进入MASH纤维化的III期临床试验),以及NR1I2、CEBPA和NR1I3(均具有肝脏代谢调节的临床前证据)(图S5H-S5N)。
为了探究驱动肝细胞应激反应的核心TFs,研究人员应用MATCHA构建了一个连接基因与TFs的双向调控网络(图5G)。在已鉴定的TFs中包括THRB(甲状腺激素受体β),其激动剂resmetirom 最近成为了美国FDA批准的首个用于治疗伴有中重度纤维化的MASH的药物。MATCHA分析了以下三者之间的调控关联:(1)THRB基序;(2)“纵向减少”和“持续下调”程序的激活;(3)发育相关程序及HCC S1 WNT程序的抑制(图5G)。作者最终选择了15个TFs(共20种异构体)进行实验验证(图S5O)。
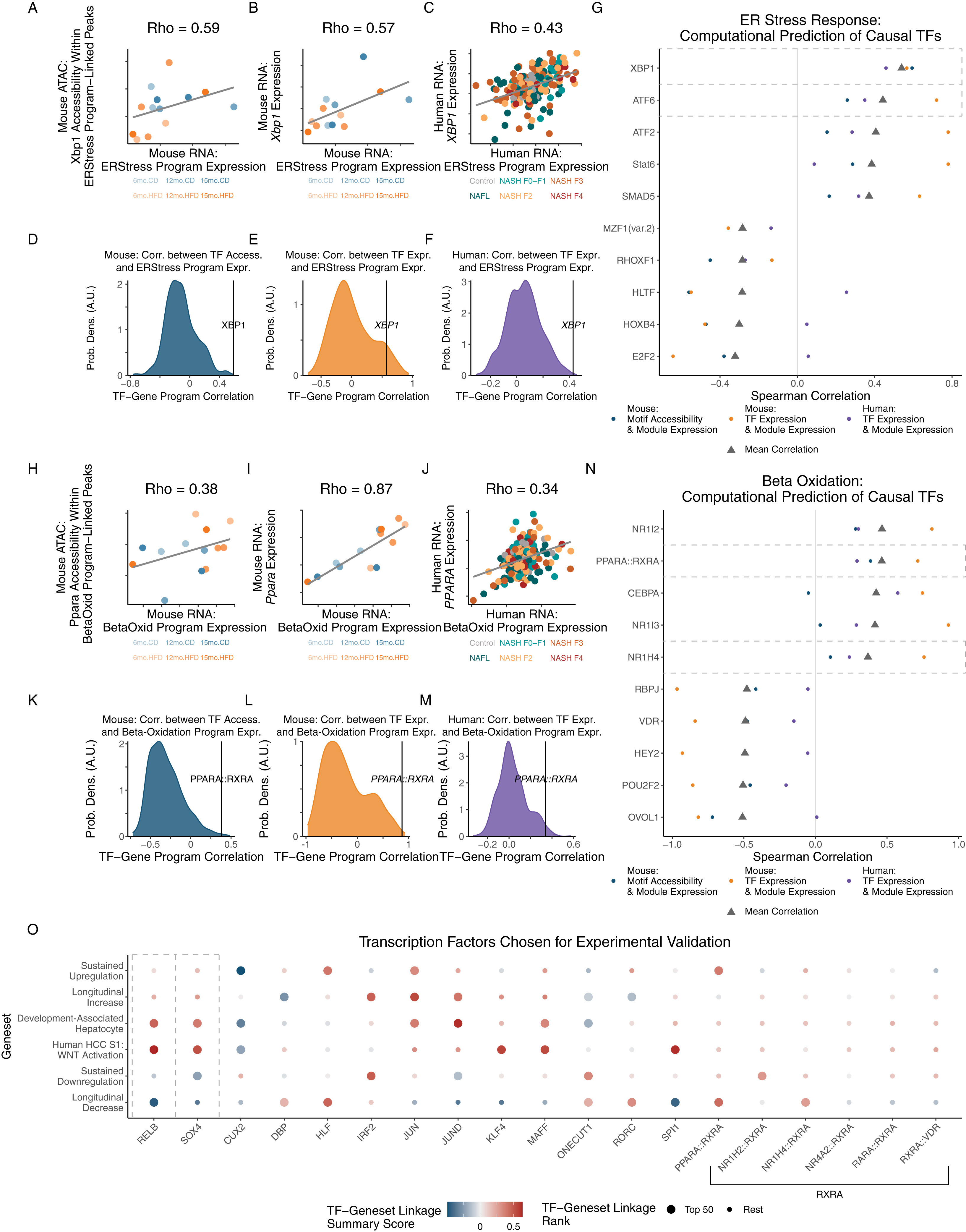
图S5. MATCHA能够对调节任意用户自定义基因程序及特定细胞表型的转录因子进行优先序分析(对应图5)
7、验证驱动应激反应且在体内提升增殖能力的细胞内源性因子
为了验证肝细胞内源性TFs对代谢应激反应的调控作用,研究人员在富含脂质的培养基中对HepG2细胞进行了人源转录因子的体外过表达实验,并结合scRNA-seq、活细胞成像和免疫荧光检测(图6A)。阴性对照(未转导或转导蓝色荧光蛋白)的细胞根据培养基条件(即脂质处理vs对照培养基)进行聚类,证明了所测得的脂质诱导的代谢应激反应具有特异性(图6B)(小编注:转导是指利用病毒作为载体,将外源基因导入细胞)。阳性对照实验证实在高脂培养基中过表达PPARA会上调其已知的靶基因,同样,RELB、JUN和RORC的过表达也驱动了代谢相关基因的表达,这与它们作为NF-κB,AP-1通路和代谢感知的效应器功能相一致,从而证明了实验方法的有效性。SOX4、CUX2和HLF下调了蛋白质分泌相关基因并上调了胎儿相关HCC标志物表达,鉴于SOX4和RELB对代谢应激相关的转录及细胞功能具有显著影响,研究人员将其作为研究重点。
SOX4和RELB的过表达均增加了“纵向增加”、“持续上调”和HCC S1特征程序,同时下调了“纵向减少”和“持续下调”程序(图6C,6D)。在功能上,SOX4和RELB虽减少了脂质积累,但增加了活性氧(ROS)与核内p53蛋白的积累。此外,SOX4还促进了细胞周期进程(以核KI67为标志),表明细胞在应激状态下仍能提升增殖能力 (图S6C-S6G)。
在人体相关性方面,SOX4和RELB的表达均随着MASLD严重程度的增加而增加,并预示着HCC患者生存期恶化(图6E-6H)。类似于在人类HCC患者中进行的一场“天然遗传扰动实验”,RELB基因的拷贝数变异(CNVs)不仅驱动了RELB自身表达的显著改变,还驱动了下游应激反应程序的协同变化(图S6H-S6P)。通过整合肝脏基因表达与癌症发病率相关联的队列数据,研究人员发现在非癌变的人类肝组织中,SOX4和RELB的高表达可提高未来HCC的发病风险,这将SOX4和RELB对非癌变肝细胞应激反应的调控,与未来HCC的发生有效地联系起来(图S6Q和S6R)。
鉴于SOX4在体外表现出的转录调控和促进增殖效应,研究人员用AAV诱导SOX4在HFD小鼠的肝脏中过表达(图6I和S6S-S6X)。通过snRNA-seq参考映射分析发现(小编注:snRNA-seq参考映射是指通过将新测序细胞的高维转录组特征投影到已建立的全景参考图谱上,实现对未知状态细胞的“定格取证”。在本研究中,该方法量化了SOX4如何驱动肝细胞发生转录轨迹偏移,从而在分子水平证明了该因子对病程演化的加速作用),有64%的SOX4过表达的肝细胞被识别为相当于HFD喂养15个月的状态,这表明SOX4在体内驱动并加速了肝细胞的转录重编程。SOX4的过表达改变了肝细胞的应激反应程序,并且其变化的方向与长达数月至数年的慢性应激过程高度一致(如上调了发育、促生存及再生相关的细胞周期调控因子,下调了代谢酶及分泌蛋白)。在体内蛋白水平及功能方面,研究发现SOX4的过表达直接增加了KI67阳性的肝细胞比例,并提升了CyclinD1(该蛋白的表达随细胞增殖而增加)的蛋白丰度(图6M-6O)。最后,SOX4的过表达增加了WNT效应蛋白β-catenin的丰度(小编注:作为WNT通路的核心信号传导子,β-catenin的丰度增加及其入核易位,是诱导肝细胞去分化及开启癌性转录程序的关键分子事件。它是连接慢性代谢应激与恶性转化的关键节点通路效应物)(图6O)。综上所述,数据表明:肝细胞内源性因子足以驱动上述转录应激反应程序,并在饮食诱导的肝脏应激微环境中增强肝细胞的增殖能力。
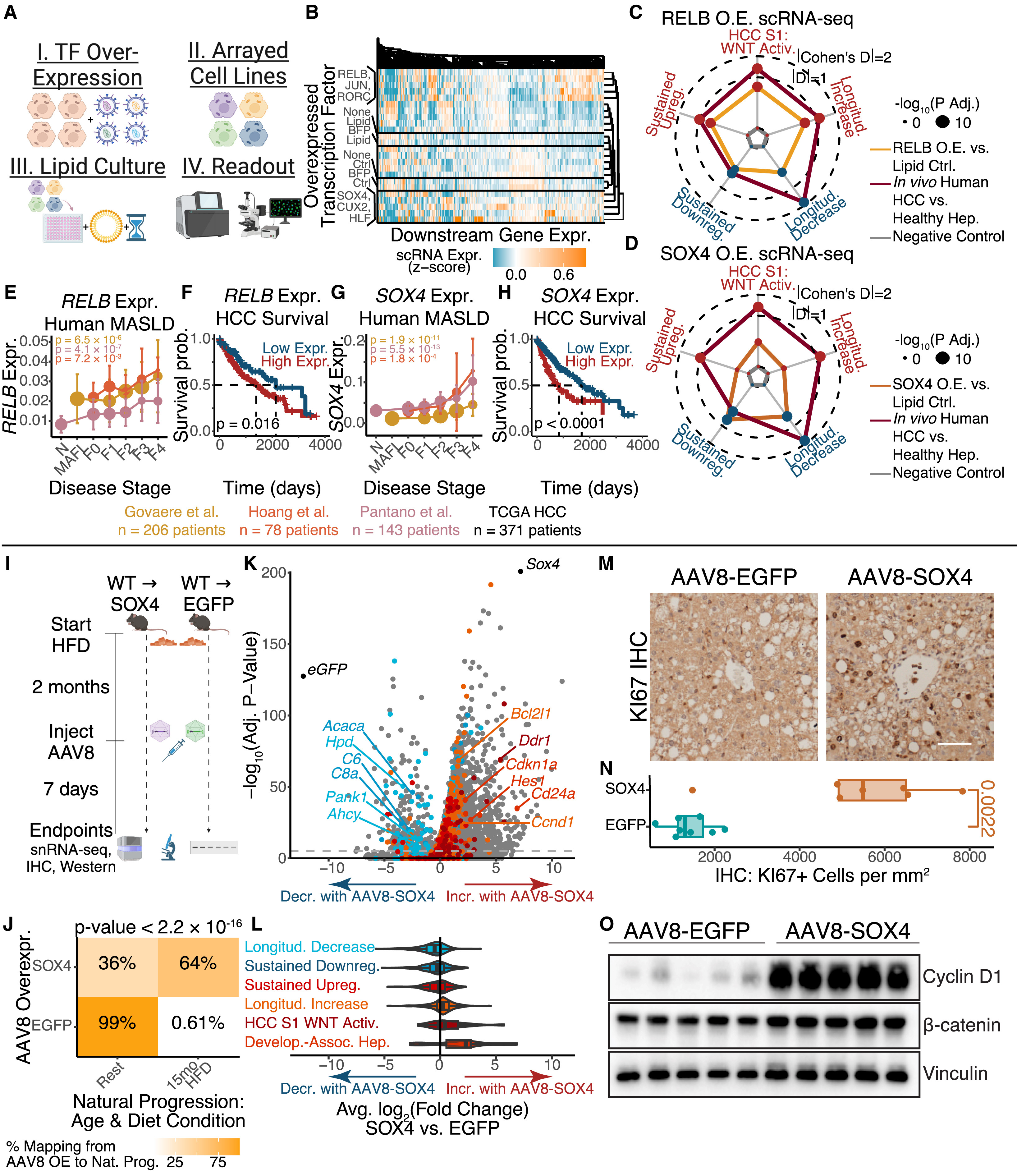
图6. 通过人类体外实验及小鼠体内遗传扰动,验证SOX4和RELB对肝细胞代谢应激反应的调节作用

图S6. RELB与SOX4对肝细胞代谢适应程序的调控作用及其与人类体内疾病进展及未来肿瘤发生关系的关联分析(对应图6)
8、人类空间转录组学鉴定肝细胞应激反应的空间结构化信号驱动因子
最后,研究人员试图揭示可能参与调控肝细胞应激反应程序的潜在环境信号,以及空间结构化的细胞间相互作用。为了将上述应激反应的发现扩展到MASH肝硬化的组织背景中,研究人员组建了一个性别平衡的人类MASH肝硬化肝组织微阵列(TMA),并进行了单细胞分辨率空间转录组学分析(共18名参与者,723,466个细胞)(图7A,7B和图S7)。
与先前跨物种多组学分析结果一致的是,人类MASH肝硬化中的肝细胞应激反应程序也呈现出方向一致的变化:人类HCC S1亚型相关的WNT通路激活,“持续上调”、“纵向增加”及发育相关的肝细胞基因表达显著增强;而“持续下调”和“纵向减少”的基因表达则显著减弱(包括HMGCS2的显著下调)(图7C;这与图3,4,6和S6中的人类转录组和蛋白质组分析结果相互补)。此外,空间数据进一步支持了MATCHA计算推断及遗传扰动实验的验证结果:相比于SOX4-和RELB-肝细胞,SOX4+和RELB+肝细胞表现出更高水平的应激增强型基因表达,以及更低水平的应激减弱型基因表达(包括HMGCS2)(图7D)。
为了探究脂肪-肝脏信号传导轴的潜在作用,研究人员发现,无论是在瘦型MASH患者还是高BMI的MASH患者中(BMI分别为26.6±0.81 vs. 35.7±0.80),肝细胞的空间应激反应同样显著(图S7E)。通过对区分肥胖与肝脏病理的小鼠模型进行荟萃分析,以及对脂质或细胞因子TGFB1处理的人类肝脏类器官进行研究,同样发现,单纯的肥胖或脂质均不足以驱动严重的代谢应激反应(图S7F-S7H)
接下来,为识别疾病相关的细胞间相互作用及多细胞枢纽,研究人员将肝细胞应激反应作为局部邻域细胞组成的函数进行了地理回归分析(小编注:地理回归是一种空间分析技术,广泛应用于地理学及涉及空间模式分析的相关学科。其通过在空间范围内每个点处建立局部回归方程,来探索研究对象在某一尺度下的空间变化及相关驱动因素,并可用于对未来结果的预测)(图7E)。结果发现,与极端肝细胞应激反应共定位最紧密的是CD9+TREM2+瘢痕相关巨噬细胞,此类细胞被认为与肝硬化中星状细胞胶原转录的增加有关(图7F)。与其相一致的是,在HFD小鼠scRNA-seq和多重免疫荧光实验中也发现了此类巨噬细胞的富集,且在疾病早期阶段即存在(图S7I-S7N)。在人类MASH肝硬化微阵列空间组学中,富集程度排在第二和第三的细胞类型分别是IGFBP1+PCK1+胆管细胞和HES1+CD36+内皮细胞,它们均与肝脏疾病反应有关。这些细胞类型的空间共定位预示着促进肝细胞应激反应的信号相互作用。在微观和介观尺度上,可以清晰观察到这三类细胞与应激肝细胞共定位形成的空间枢纽(图7G)(小编注:“介观尺度” 指的是介于微观(单个细胞或亚细胞结构)和宏观(整个组织或器官)之间的观察尺度。它关注的是由多个细胞聚集、相互作用所形成的空间组织结构。)。
为了揭示空间结构化的信号调节网络,研究人员将每个肝细胞基因的表达水平作为其局部邻域内分泌信号蛋白转录本的函数,进行了地理回归分析(图7H)。该方法成功复现了肝小叶门脉周围-中央静脉周围的空间结构化WNT基因调节环路:其中,在中央静脉周围富集的WNT配体是:(1)驱动已知中央静脉标志物及经典WNT靶基因最强的正向信号(2)抑制门静脉周围标志物最强的负向信号(图7I)。LTB、JAG1和IL32是协同正向调节持续上调和纵向增加基因程序(包括去分化标志物、细胞死亡保护基因及损伤相关下游信号)的关键信号。其中,LTB可通过RELB激活非经典NF-κB信号通路,JAG1介导的Notch信号则促进肝纤维化,而循环系统中的IL32已被确认为MASLD的生物标志物。RELB与SOX4本身呈现出共聚类特征,并表现出与上述基因程序相似的上游信号调节模式,这进一步证明了这些TFs与肝细胞应激反应的紧密联系。
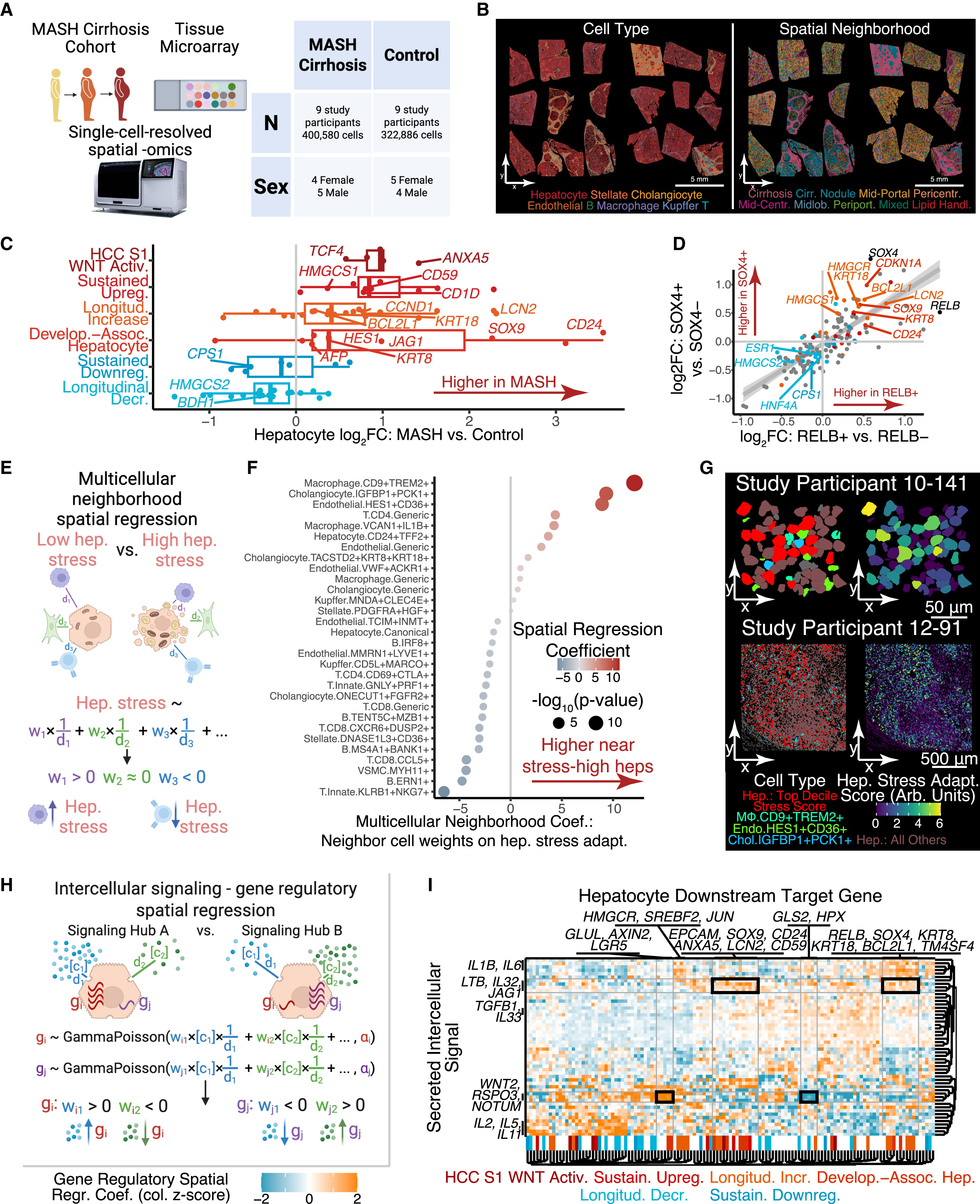
图7. 人类MASH空间转录组图谱揭示了肝细胞应激反应的多细胞信号转导背景
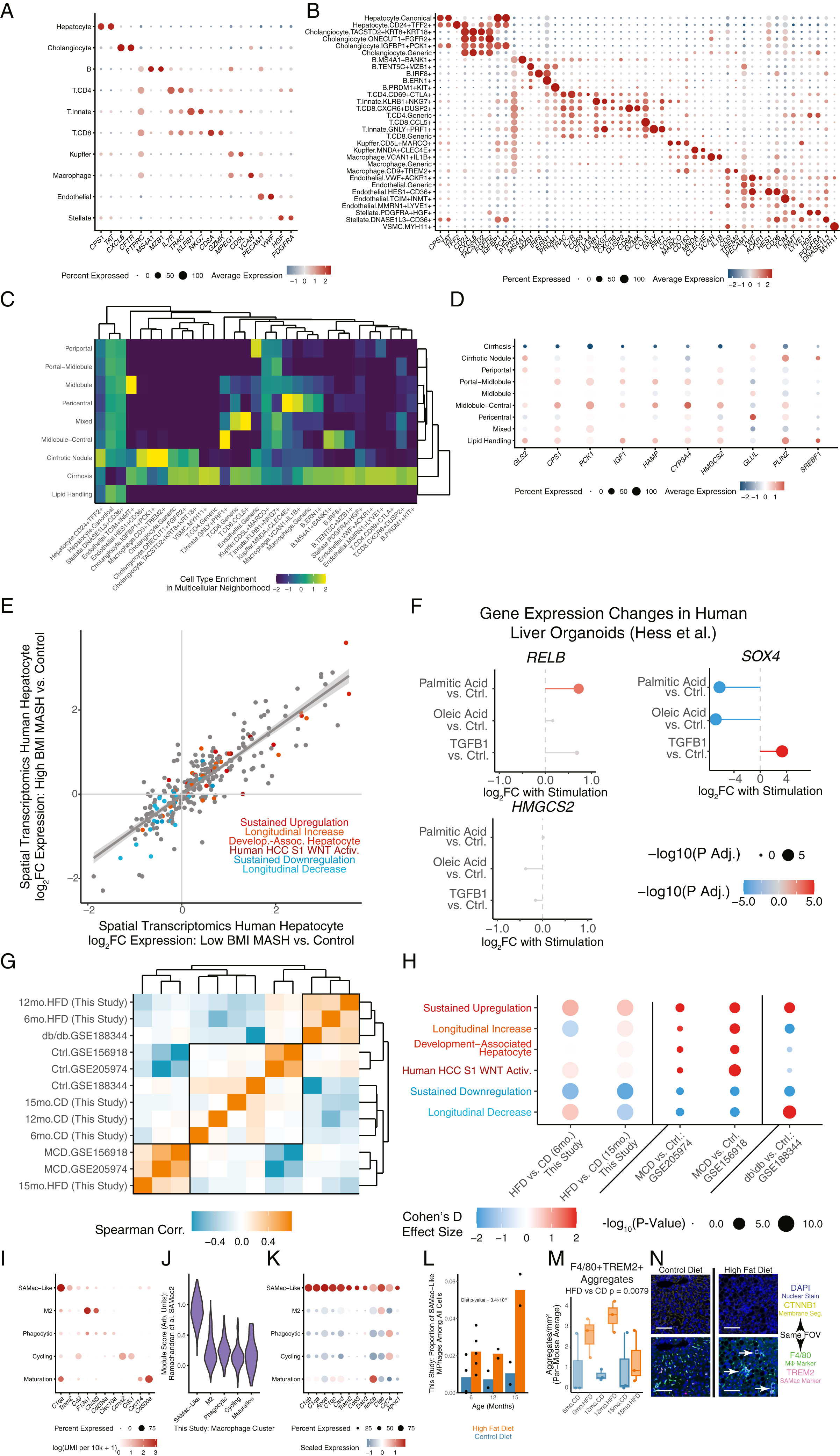
图S7. 将肝细胞应激反应置于人类肝脏空间组织、人类BMI、脂质内源性反应、炎症与肥胖的对比及免疫效应等多重语境下的关联分析(对应图7)
总结
本研究通过跨物种组学分析与空间转录组学技术,揭示了肝细胞在慢性代谢应激下向肝细胞癌(HCC)演变的分子机制。长期高脂饮食(HFD)在肝脏内诱导形成由瘢痕相关巨噬细胞等构成的多细胞应激枢纽,这些细胞通过释放LTB、IL32和JAG1等环境信号,激活肝细胞内空间结构化的信号环路。在这些外源信号驱动下,肝细胞发生转录重编程:激活转录因子SOX4与RELB,上调发育相关程序、促生存信号及Cyclin D1等细胞周期调节因子,并激活WNT/β-catenin通路,同时下调HNF4A和HMGCS2等维持肝细胞代谢的关键因子。这种从“代谢执行”向“病理性再生”的身份切换,使得受损肝细胞在脂毒性微环境中获得生存与增殖优势,从而显著提升了癌症发生风险。总的来说,研究揭示了“慢性代谢应激-多细胞空间枢纽-转录因子(SOX4/RELB)-去分化与异常增殖”这一促癌轴,为代谢相关肝癌的早期干预提供了新思路。
原文链接:https://pubmed.ncbi.nlm.nih.gov/41435820/
https://blog.sciencenet.cn/blog-3483272-1523850.html
上一篇:Cell Metabolism:巨噬细胞PD-1:代谢能量新开关
下一篇:Science:米色脂肪 “丢身份”,血管血压闹脾气!
