博文
Cell Metabolism:剪刀手,爱胰腺
||
代谢学人
Cell metabolism:剪刀手,爱胰腺
撰文 | 郭钰涵 李姿萱 李悦 周文豪 邱瑾
编辑 | 孟美瑶
校对 | 李姿萱

背景介绍
HNF1A编码转录因子肝细胞核因子-1α,是孟德尔糖尿病中最常见的突变基因(小编注:HNF1A主要在人体的肝脏、胰腺、肾脏和消化道等器官中表达。在胰腺中,作为胰腺β细胞功能的核心调控者,它调控一系列与胰岛素分泌和葡萄糖代谢相关基因的表达。孟德尔糖尿病指的是由单个基因的突变引起、并且遵循孟德尔遗传定律的糖尿病类型)。HNF1A功能丧失性突变不仅会导致青少年发病的成年型糖尿病3型(maturity-onset diabetes of the young type 3,MODY3,单基因糖尿病最常见的形式),还会导致多基因2型糖尿病(T2D)的患病风险增加,而功能获得性突变可预防T2D。此外,有研究表明T2D患者的胰岛β细胞中存在HNF1A依赖性转录缺陷现象。
尽管 HNF1A在单基因和多基因糖尿病的发病机制中发挥着关键作用,但它具体在哪类细胞中调控葡萄糖稳态仍不清楚。在MODY3以及Hnf1a纯合突变小鼠中胰腺β细胞功能障碍是主要表现。HNF1A能够在肝细胞、肾脏和肠道细胞以及胰腺腺泡和胰岛细胞中表达,所有这些细胞类型都会影响葡萄糖稳态,并间接影响β细胞功能。因此,无法确定HNF1A缺陷型糖尿病是否是由于β细胞的自主缺陷(小编注:细胞自主缺陷指的是:一个细胞的功能异常或死亡,是由它自身内部的基因或分子问题导致的,与它周围的环境(如其他细胞、血液中的激素或信号)是否正常无关)。
此外,HNF1A缺陷型糖尿病的分子机制尚不清楚。HNF1A突变型β细胞表现为多种细胞缺陷,包括葡萄糖摄取缺陷,糖酵解信号传导缺陷,或线粒体呼吸缺陷,以及异常钙信号传导。但这些细胞表型并未明确HNF1A的直接靶基因,这些缺陷会带来什么生理影响仍不清楚。
近期,一篇发表在Cell metabolism上的题目为“HNF1A and A1CF coordinate a beta cell transcription-splicing axis that is disrupted in type 2 diabetes”的文章发现,HNF1A缺陷型糖尿病主要源于胰腺β细胞中HNF1A功能缺陷,而不是其他表达HNF1A的细胞类型。作者进一步确定了一个进化上保守的由 HNF1A调控的β细胞转录程序,并表明编码APOBEC1互补因子的A1CF基因是重要的HNF1A靶基因。作者证明 A1CF控制着一种以前未被识别的β细胞剪接程序,并使用单细胞基因组学和人类遗传学将该途径与T2D的分子病理生理学联系起来。总的来说,这些发现揭示了协调β细胞转录和剪接的分层机制,揭示了这两个基因调控层如何相互作用以控制细胞特异性表达,并提出这种β细胞分子途径可以用于靶向治疗。

敲黑板啦!
1. HNF1A缺陷型糖尿病的主要诱因为β细胞自主缺陷
2. HNF1A和A1CF协调β细胞转录和剪接程序
3. T2D患者表现出HNF1A-A1CF轴抑制的β细胞增加
4. 胰岛A1CF表达降低与血糖升高和T2D风险增加有关
研究结果
1、HNF1A缺陷型糖尿病是β细胞自主功能障碍的结果
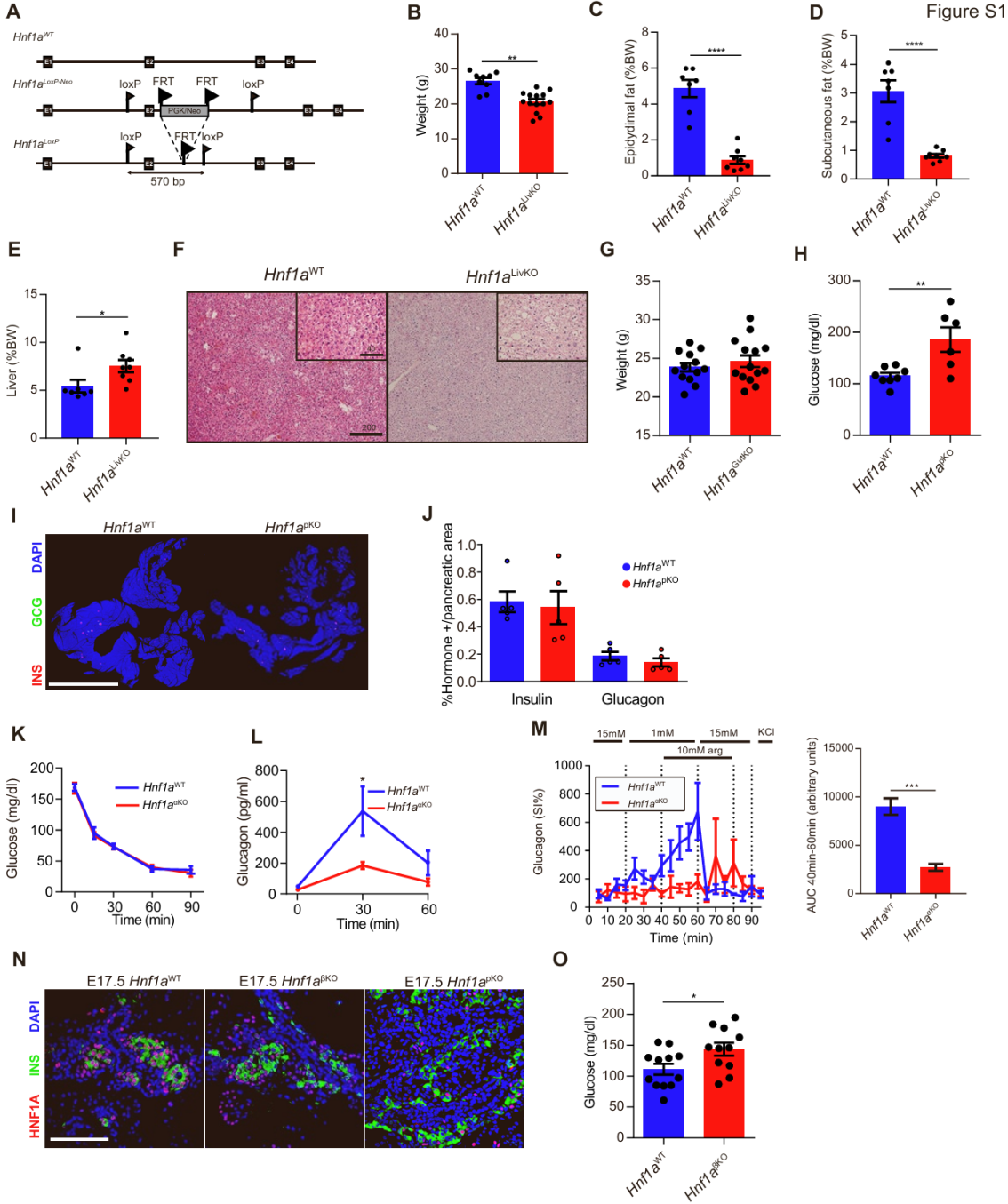
为了系统地筛选HNF1A发挥维持葡萄糖稳态作用的细胞类型,研究人员使用了loxp-cre系统构建了肝脏、肠道或胰腺特异性敲除Hnf1a的小鼠(图S1A)。
肝细胞在维持葡萄糖稳态中发挥重要功能,可以间接影响β细胞功能或质量。研究人员使用肝脏特异性Cre小鼠(Albafp-Cre,以下简称Alb-Cre)(图1A)敲除了肝细胞中的Hnf1a(Hnf1aLivKO),观察到雄性Hnf1aLivKO小鼠中重现了Hnf1a−/−小鼠的异常表型(小编注:之前有研究发现,Hnf1a全身敲除(Hnf1a−/−)小鼠患有2型糖尿病、侏儒症、肝功能障碍和高胆甾血症,存在胆汁酸转运缺陷,胆汁酸和肝脏胆固醇合成增加,HDL代谢受损。参考文献:Shih DQ, Bussen M, Sehayek E, et al. Hepatocyte nuclear factor-1alpha is an essential regulator of bile acid and plasma cholesterol metabolism. Nat Genet. 2001),包括体重和脂肪量减少,以及脂肪肝(图S1B–S1F),在之前的研究中,Hnf1a−/−小鼠的体重和脂肪量减少被归因于肝脏Igf1(胰岛素样生长因子1)表达降低(参考文献:Lee YH, Sauer B, Gonzalez FJ. Laron dwarfism and non-insulin-dependent diabetes mellitus in the Hnf-1alpha knockout mouse. Mol Cell Biol. 1998),与 Hnf1a−/−小鼠不同的是,Hnf1aLivKO小鼠没有表现出高血糖,而腹腔(ip)葡萄糖耐量(Hnf1aLivKO小鼠 272 ± 15 mg / dL vs. Hnf1aWT小鼠361 ± 12 mg / dL)被显著改善(图1B和1C),这可能是由于脂肪减少导致的胰岛素增敏作用,或者Hnf1a−/−小鼠本身肝脏葡萄糖代谢存在异常。因此,肝脏中HNF1A的缺乏不是导致在Hnf1a种系突变小鼠中观察到高血糖的原因。
肠道细胞在葡萄糖稳态调节具有重要作用,这主要归因于肠上皮细胞的糖异生能力以及肠内分泌细胞产生肠促胰岛素(小编注:肠内分泌细胞分泌的胰高血糖素样肽1(GLP1)和肠促胰岛素(GIP,也称为葡萄糖依赖性促胰岛素多肽),这两种激素在食物摄入后释放,并且可增强葡萄糖依赖性的胰岛素分泌:小肠上皮细胞对葡萄糖的吸收是由小肠刷状缘中的钠葡萄糖协同转运蛋白(SGLT1)驱动的,导致血浆葡萄糖浓度升高。血浆葡萄糖浓度的升高会引发胰腺β细胞中细胞质Ca2+ 浓度升高,从而导致胰岛素分泌增加。葡萄糖通过 SGLT1 穿过肠内分泌K细胞或L细胞的顶膜,分别触发GIP和GLP1的分泌。胰腺β细胞上的GIP受体(GIPR)和 GLP1(GLP1R)受体通过 Gs 蛋白与 cAMP 水平升高相偶联,这两种受体在血浆葡萄糖浓度升高的同时被激活,增强胰岛素的分泌) ,这类激素能调控β细胞的功能和质量。研究人员使用Vil1-Cre工具鼠切除了肠上皮细胞中的 Hnf1a,包括产生GLP1(glucagon-like peptide-1,胰高糖素样肽-1,是一种主要由肠道L细胞分泌的激素,由进食刺激触发分泌,属于肠促胰素。 GLP-1是引起的胰岛素分泌的主要激素)的细胞(Hnf1aGutKO) (图1D)。与仅表达Cre的同窝小鼠相比,Hnf1aGutKO小鼠在16周时的体重、空腹血糖、口服葡萄糖耐量或胰岛素敏感性没有差异(图1E,1F和S1G)。因此,肠道中的HNF1A缺乏并不能重现Hnf1a−/−小鼠的糖尿病表型。
拓展阅读
肠上皮细胞糖异生
肠上皮细胞糖异生(Intestinal Gluconeogenesis, IGN)是指小肠上皮细胞利用非碳水化合物前体(如短链脂肪酸、氨基酸等)合成葡萄糖的过程。
(1)激活条件:膳食纤维有两个主要成分:不溶性纤维(主要是纤维素和木质素)和可溶性纤维(如低聚半乳糖和低聚果糖),它们能够被肠道微生物群发酵成短链脂肪酸(SCFA)乙酸盐、丙酸盐和丁酸盐,并通过互补机制激活 IGN:①丙酸本身能够作为IGN的底物,通过激活肠上皮细胞上的脂肪酸受体FFAR3,引发肠-脑神经信号传导,从而上调IGN相关基因的表达。②丁酸(Butyrate):它主要通过cAMP依赖机制来激活IGN相关基因的表达。
(2)与肝脏糖异生的区别:
肝脏糖异生(HGN)用来维持血糖稳定,在饥饿状态下为大脑、红细胞等提供必需的葡萄糖,葡萄糖直接释放到肝静脉,进入体循环,直接影响全身血糖水平,主要受胰高血糖素和胰岛素的直接调控。
IGN释放的葡萄糖能够作为一种信号分子,会被肠道内门静脉葡萄糖传感器检测到,该传感器通过外周神经系统将其信号传输到大脑,提高外周组织(如肝脏、肌肉)对胰岛素的敏感性,并抑制肝脏的糖异生作用,帮助降低血糖水平。
参考文献:
[1] De Vadder F, Kovatcheva-Datchary P, Goncalves D, et al. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell. 2014
[2] Delaere F, Duchampt A, Mounien L, et al. The role of sodium-coupled glucose co-transporter 3 in the satiety effect of portal glucose sensing. Mol Metab. 2012
为了敲除胰腺细胞中的Hnf1a,研究人员首先使用了Pdx1-Cre工具鼠,它在所有胰腺上皮细胞的前体细胞中都具有活性(Hnf1apKO) (图1G)。与仅表达Cre的同窝小鼠相比,Hnf1apKO小鼠在断奶时表现出高血糖(图S1H),并在13周时表现出空腹和餐后高血糖(405 ± 67 mg / dL),餐后血浆胰岛素降低(Hnf1apKO 0.09 ± 0.01 vs. Hnf1aWT 0.28 ± 0.06 ng / mL)(图1H和1I)。然而,Hnf1apKO小鼠的胰腺除了在部分小鼠中的脂肪浸润斑之外,形态没有发生变化,并且相对β细胞面积(β细胞/胰腺面积分数)没有显著差异,表明该模型中的糖尿病主要是由β细胞功能障碍引起的,而不是β细胞数量减少(图S1I和S1J)。胰腺α细胞通过旁分泌机制调节β细胞功能,而MODY3患者对高血糖刺激具有异常的胰高血糖素反应。研究人员因此采用α细胞特异性诱导型Cre品系(Gcg-CreERT2),在围产期特异性敲除α细胞中的Hnf1a基因(以下简称Hnf1aαKO小鼠)(图1J)(小编注:“围产期”指小鼠快出生或刚出生后不久的这段时间)。将小鼠饲养至12周龄时,Hnf1aαKO小鼠口服葡萄糖后的血糖水平和胰岛素分泌水平与他莫昔芬诱导的Cre阴性同窝对照组无显著差异(图1K、1L)。然而,在体内实验中,Hnf1aαKO小鼠对胰岛素诱导低血糖的胰高血糖素反应减弱;体外实验中也观察到其对低葡萄糖和精氨酸刺激下的胰高血糖素应答减弱(图S1K-S1M)。由此可见,α细胞Hnf1a缺失虽不导致糖尿病,但可能影响反调节激素应答机制。
最后,研究人员采用Ins1-Cre敲入等位基因,在妊娠晚期β细胞形成后逐渐灭活其Hnf1a基因,最终实现成年β细胞中Hnf1a的近乎完全缺失(Hnf1aβKO)(图1M和S1N)。与Cre的同窝对照组小鼠相比,Hnf1aβKO小鼠在离乳期即表现为高血糖(图S1O);16周龄时表现为腹腔葡萄糖耐量异常(60分钟血糖值336±13 mg/dL vs. 256±19 mg/dL)及胰岛素分泌减弱(15分钟胰岛素值0.04±0.03 ng/mL vs. 0.27±0.04 ng/mL)(图1N和1O)。由此可见,β细胞特异性Hnf1a缺失能成功重现Hnf1a−/−小鼠中的高血糖表型,表明HNF1A的β细胞自主性功能对糖尿病发生具有关键作用。
图S1. 对携带细胞特异性Hnf1a基因失活的小鼠进行研究,相关结果见图1
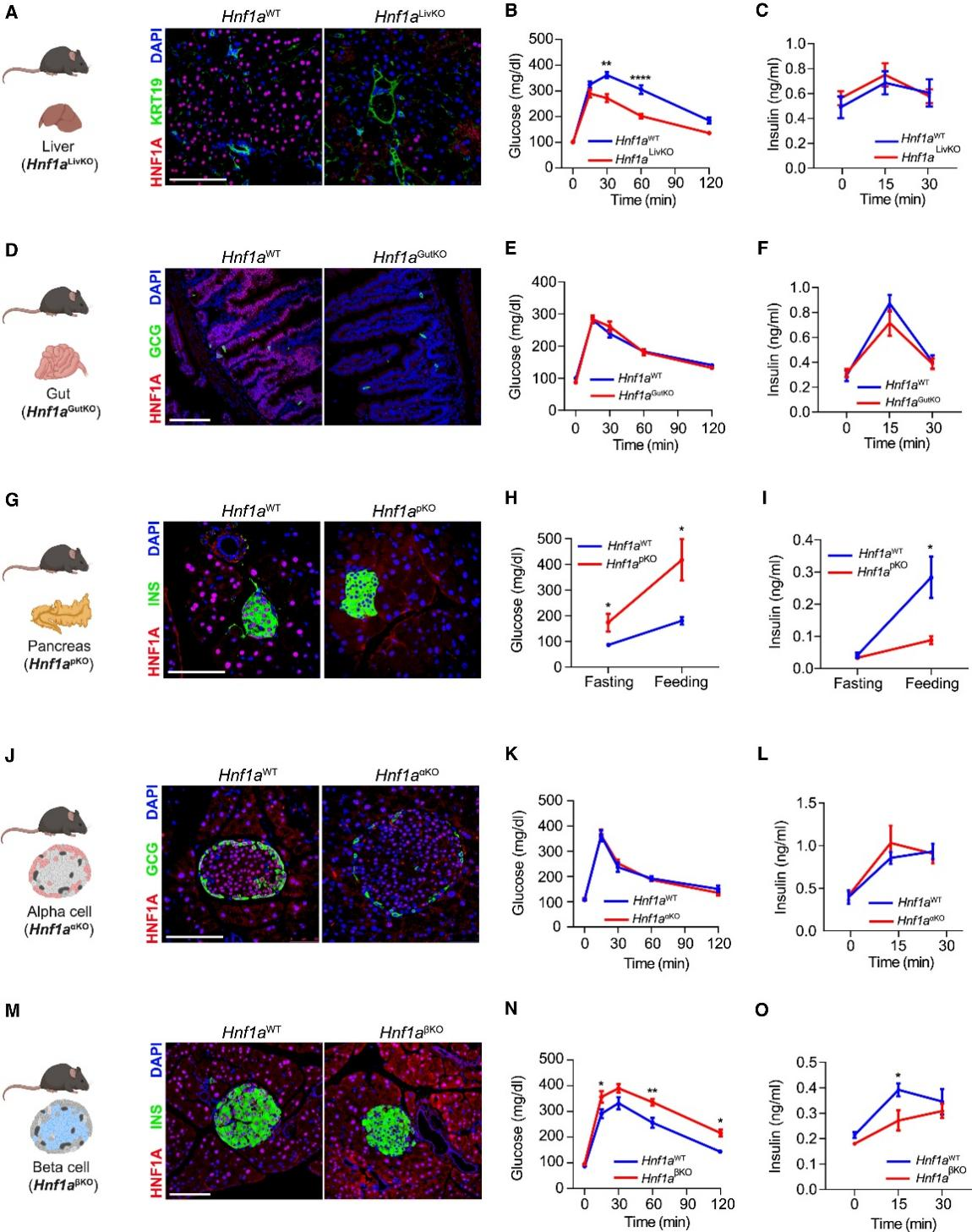
图1. HNF1A依赖型糖尿病需要β细胞中该基因的缺失
2、HNF1A调节小鼠和人β细胞中的保守转录程序
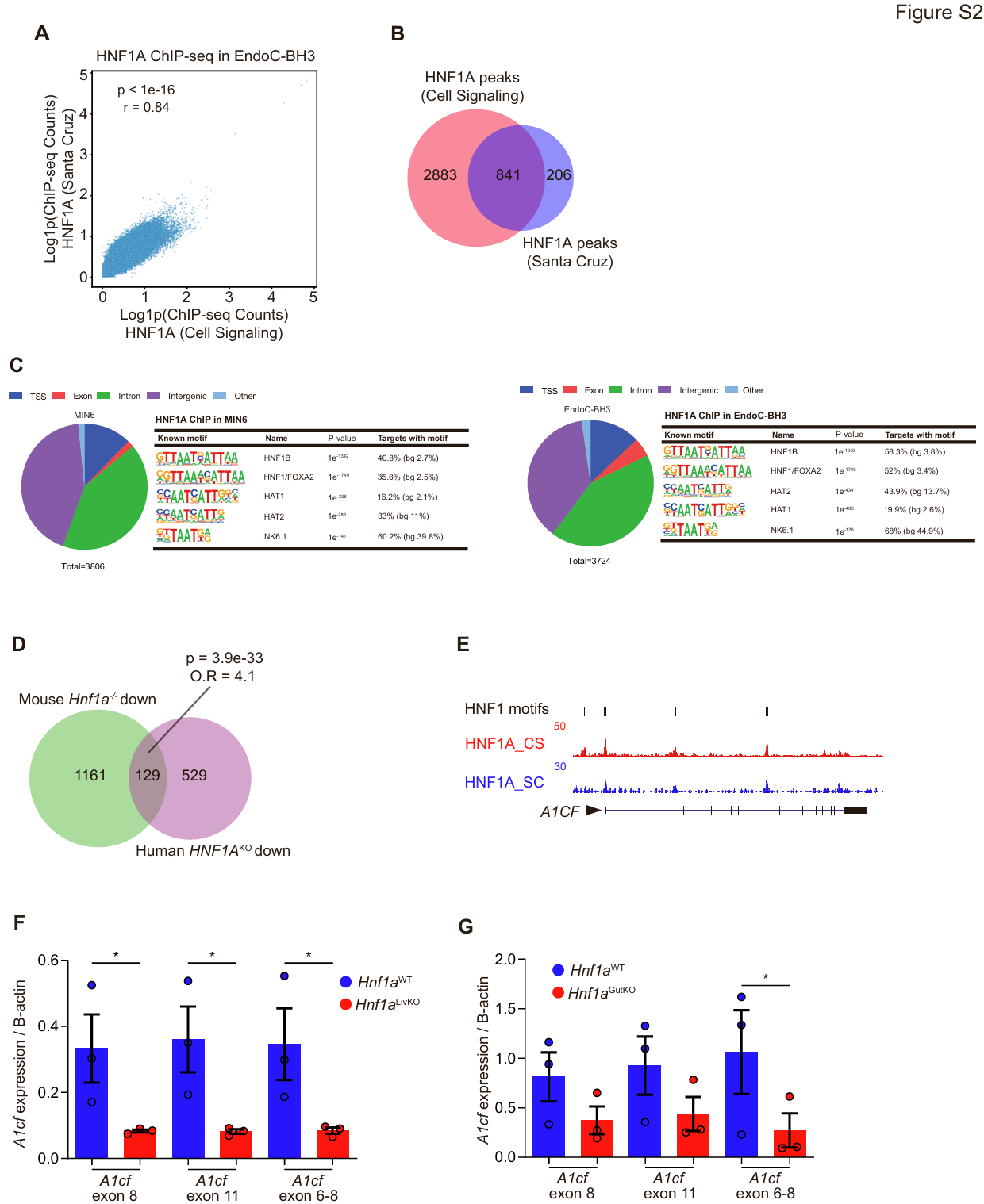
接下来,研究人员进一步探究了HNF1A在β细胞中的分子功能。鉴于小鼠纯合子与人类杂合子HNF1A突变均会导致糖尿病,研究人员试图在小鼠和人类β细胞中鉴定保守的HNF1A靶基因。为此,研究人员检测了携带双等位基因HNF1A缺失的人EndoC-βH3 β细胞及成年Hnf1a⁻/⁻小鼠胰岛。正如之前对HNF1A缺失胰岛细胞的研究所预期的那样,这两种模型的转录组均存在显著差异(图2A、2C)。为确定HNF1A的直接效应,研究人员在人EndoC-βH3和小鼠MIN6 β细胞中进行了HNF1A染色质免疫沉淀测序(ChIP-seq)(图S2A-S2C)(小编注:图S2A:使用了Cell Signaling Technology和Santa Cruz两种品牌的HNF1A抗体,在人EndoC-BH3 β 细胞中进行ChIP-seq,结果显示:两种HNF1A抗体检测到的结合位点信号强度一致性很高。图S2B:两种抗体ChIP-seq结果进行merge,显示Cell Signaling Technology的结合效率更高(可包含Santa Cruz抗体ChIP-seq的80%以上);故图S2C:使用 Cell Signaling Technology抗体ChIP-seq结果分析在小鼠MIN 6 β细胞和人EndoC-BH3 β细胞中的HNF 1A结合位点。饼图展示了HNF1A结合区域在基因组不同区域(转录起始位点(TSS)、内含子、外显子、基因间区)的分布比例,右边的表格列出了HNF1A结合位点中富集的DNA基序(Known motif),包括与HNF1家族(如HNF1B、HNF1/FOXA2)及β细胞关键因子(如 NK6.1)相关的序列,提示HNF1A可能通过结合这些序列,调控下游基因表达)。发现HNF1A结合基因在人与小鼠突变模型下调的基因中显著富集(p值分别为1.9×10⁻¹³和3.4×10⁻¹¹)(图2A-2D)(小编注:O.R.通常指比值比或优势比(Odds Ratio),在统计学中,是用于衡量两组事件发生优势的相对差异的指标;在生物学中,是常用来衡量暴露因素与疾病关联强度的统计指标,指病例组中暴露人数与非暴露人数的比值除以对照组中暴露人数与非暴露人数的比值。O.R.=1表示该因素对疾病的发生无影响,O.R.>1:表示暴露因素与疾病呈正相关,该因素是危险因素,O.R.<1:表示暴露因素与疾病呈负相关,该因素是保护因素。在这里,它应该是用于比较 “HNF1A结合的基因” 与 “非HNF1A 结合的基因” 的差异。若 O.R. > 1:说明HNF1A 结合的基因更倾向于发生该类表达变化(如图2B 中 “Down” 组 O.R.=2.1,代表HNF1A结合的基因更倾向于表达下调。这与HNF1A在胰腺β细胞作为转录激活因子发挥主要作用的观点一致。图S2D中,小鼠与人类中HNF1A敲除后,与HNF1A结合的靶基因中下调基因的重叠情况,有129个基因在小鼠和人类中都下调,并且OR=4.1表明HNF1A敲除后表达下调的事件概率是不敲除组的4.1倍)研究人员发现,在人类和小鼠HNF1A缺陷模型中下调直系同源基因存在显著重叠(图S2D)。更重要的是,直接靶基因多数表现为下调(图2E和2F)。据此研究人员确定了129个在人类和小鼠模型中共同下调的核心直系同源基因(图S2D)(小编注:这里的129个基因是rna-seq表达下调,人鼠overlap之后得到的。直系同源基因指的是在不同物种中源自同一祖先基因,具有相似功能。在此处,“核心直系同源基因”是指那些在人和小鼠中,HNF1A缺失后表达下调的基因,具有高度功能保守性。其p值是通过Fisher's exact test(费希尔精确检验)计算得出的,用于评估人和小鼠中HNF1A直接靶标且下调的基因是否显著重叠。Fisher精确检验的计算方法如下:对图S2D来说,有四个参数:a = 129 (人和小鼠重叠的基因数),b = 529 (人特有,小鼠没有的基因数),c = 1161 (小鼠特有,人没有的基因数),d = 13,181 (小鼠和人都没有的基因数)。行1总和 (a+b) = 658 (人靶标基因总数),行2总和 (c+d) = 14,342 (非人靶标基因总数),列1总和 (a+c) = 1290 (小鼠靶标基因总数),列2总和 (b+d) = 13,710 (非小鼠靶标基因总数),总和 (N) = 15,000 (背景基因总数)。零假设 (H₀) 是:两个物种的靶标基因列表是独立的,没有关联。重叠的129个基因纯粹是随机发生的。假设 (H₁) 是:两个列表存在正相关(重叠比随机预期更多)。计算出来p<0.05代表零假设不成立,得出结论:人和小鼠的HNF1A靶标基因列表的重叠程度在统计学上极其显著,远超随机预期。这支持了HNF1A调控通路在进化上保守的结论)。其中许多直系同源基因对编码潜在相关的β细胞功能,例如内质网(ER)应激,氧化应激,脂质代谢和RNA剪接(图2G和2H)。值得注意的是,RNA结合功能是在两个物种下调基因中最显著富集的类别之一(图2I)。
研究人员还探究了HNF1A在天然人类β细胞中的调控作用。研究人员推测,单细胞水平上HNF1A mRNA的变异会导致其调控转录本相关的表达差异,并基于ESPACE人类胰腺细胞图谱联盟提供的人类β细胞转录组数据对此进行了验证。研究人员聚焦于107个保守的HNF1A靶基因(小编注:这里的107个靶基因是从129个基因中进一步筛选得出的。如后文所述,129 个基因中,只保留了那些在人类胰岛β细胞单细胞数据(VASA-seq)中表达率超过5%的基因(即在超过5%的β细胞中可检测到表达)。进一步的筛选是为了提高分析的可靠性),这些基因在>5%的人β细胞中表达,并计算了它们与HNF1A mRNA在β细胞间的两两相关性。与表达水平匹配的对照组基因相比,这107个基因与HNF1A mRNA水平显示出明显更高的相关性(图2J;HNF1A dep.genes组)。而被HNF1A直接结合的靶基因与HNF1A mRNA水平在两个物种中均表现出更强的相关性(图2J; HNF1A dep.+bound genes组)(小编注:这里又将rna-seq overlaop得到129个基因进一步与hnf1a直接结合基因 overlaop后得到32个基因,为在人和鼠的细胞中既下调又能与hnf1a直接结合的基因;并与HNF1A mRNA水平相关性最强)。值得注意的是,在HNF1A突变体中下调最显著的基因,在原代β细胞中也显示出最高的相关性数值,包括已知的HNF1A靶基因如HNF4A(在18,242个基因中排名第2)、HASTER(HNF1A-AS1,排名第19)和A1CF(排名第6)(图2K)。因此,共表达分析证实HNF1A在天然人类β细胞中调控着相同的保守基因程序。总之,这些发现揭示了小鼠和人类β细胞中由HNF1A调控的保守转录程序,为理解HNF1A缺陷型糖尿病的分子机制提供了支持。
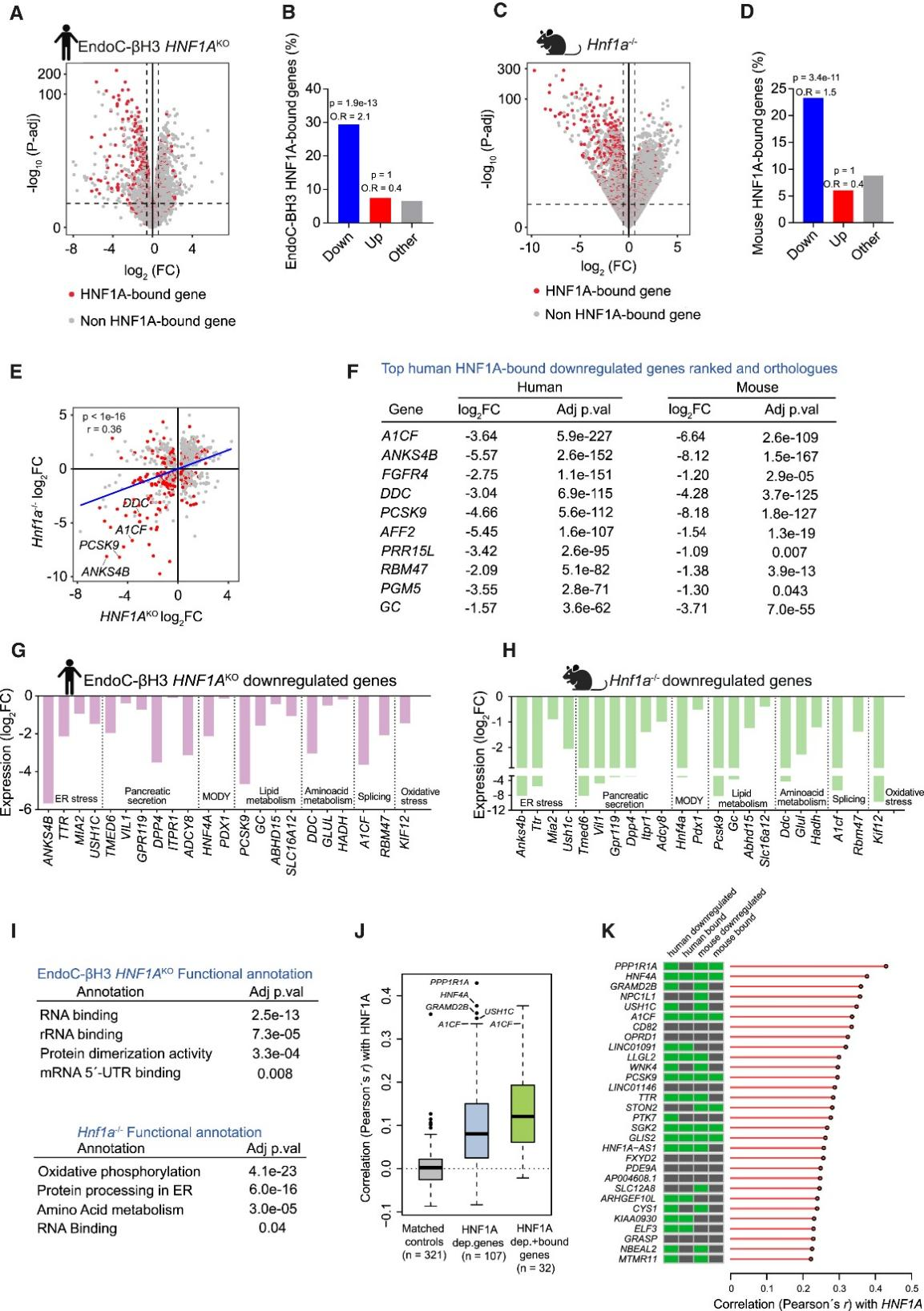
图2.HNF1A调节小鼠和人β细胞中的保守转录程序
图S2. 小鼠与人类β细胞中HNF1A的调控机制
3、HNF1A缺乏导致RBP基因表达降低
研究人员通过进一步分析发现,编码RNA结合蛋白(RBPs)的转录本在下调基因中显著富集(图2F-2I),其中包括5种在小鼠和人类HNF1A敲除模型中均下调的RBP直系同源蛋白(图3A-3C)。特别值得注意的是,编码APOBEC1互补因子的A1CF基因,在小鼠和人类HNF1A敲除模型中表达水平极低(图2F、3B、3C),且在天然β细胞中与HNF1A mRNA的相关性位居前列(图2J、2K)。小鼠和人类的A1CF基因均存在多个HNF1A结合位点,表明其受到直接调控(图3D、3E、S2E)。免疫定位研究显示,A1CF在大多数小鼠胰岛细胞中均有表达,而在Hnf1a突变体中其表达显著降低(图3F)。
A1CF最初被报道为APOBEC1 RNA编辑复合物的辅助因子,但后续研究发现其具有APOBEC1依赖性RNA编辑活性;最新研究表明它是编码果糖和甘油代谢酶的肝脏转录本选择性剪接的直接调控因子。值得注意的是,A1CF mRNA仅在表达高水平HNF1A mRNA的组织中特异性表达,包括肝脏、胃、肠道、肾脏和胰腺(图3G),且在组织特异性Hnf1a突变体小鼠的肝脏和肠道中,A1cf mRNA表达也呈现部分下降(图S2F和S2G)。这些发现表明,HNF1A是β细胞中A1CF的关键调控因子,并提示HNF1A可能间接影响细胞特异性的RNA加工过程。
4、HNF1A控制小鼠和人β细胞中的选择性剪接程序
为探究HNF1A对A1CF的调控是否影响β细胞的RNA剪接,研究人员通过复式多变量转录本剪接分析(rMATS)方法,对小鼠和人类HNF1A缺陷模型的RNA测序(RNA-seq)数据进行了分析。研究发现,HNF1A缺陷不仅导致β细胞发生重大转录本变化,还会明显改变RNA剪接模式(图4A-4D)。研究人员在HNF1A缺陷小鼠胰岛和人类EndoC-βH3细胞中分别鉴定出864和2,362个差异剪接事件,其中约60%-70%为外显子包含或跳跃事件(图4B和4D)(小编注:外显子包含/跳跃是可变剪接最直接的形式。指的是对于某一个特定的外显子,剪接体可以做出两种选择:①外显子包含(EI):将这个外显子保留在最终的成熟mRNA中。②外显子跳跃(ES):将这个外显子完全剔除,不包含在最终的成熟mRNA中)。重要的是,大多数差异剪接基因并未显示mRNA表达水平的差异(图S3A和S3B)(小编注:即火山图中灰色标注的部分,占比为大多数)。
为评估HNF1A依赖性RNA剪接程序的保守性,研究人员计算了ΔPsi值(差异剪接包含百分比),以量化突变组与对照组中包含差异剪接外显子的转录本百分比(小编注:ΔPsi是剪接变化量的定量指标。它衡量的是一个特定的外显子或其它剪接事件在不同组别下,被包含到成熟mRNA中的比例差异。简单来说,ΔPsi数值的大小直接反映了剪接方式改变的剧烈程度)。分析显示,人类与小鼠的HNF1A依赖性剪接变化呈显著正相关(图4E)。研究人员在两个物种中鉴定出共有134个同源外显子的剪接受到HNF1A依赖性剪接的调控(图4F)(小编注:如前所述:p值是通过Fisher's exact test(费希尔精确检验)计算得出的,用于评估人和小鼠中HNF1A依赖性剪接的靶标是否显著重叠。p<0.05的意义是: 该134 个受到HNF1A依赖性剪接的重叠事件的概率在统计学上极其显著,显著高于随机重叠的概率)。在差异剪接最显著的五个基因中,SLC7A2是参与精氨酸诱导胰岛素分泌的阳离子氨基酸转运体,SEC31A是COPII包被蛋白,介导β细胞中胰岛素原加工和内质网囊泡出芽,而MYO6则与分泌通路中的膜运输过程相关(图4G-4M)。
与HNF1A缺陷细胞中A1CF mRNA减少的结果一致,在HNF1AKO细胞中发生的互斥差异性剪接事件(MXE)在其侧翼序列中富集了A1CF识别序列(图S3C)。(小编注:“侧翼序列”指的是与某个特定核心区域或位点直接相邻的DNA或RNA序列。在此处,“侧翼序列是指与发生“差异性剪接”的外显子直接相邻的内含子区域);而在Hnf1a⁻/⁻胰岛中发现的差异性剪接事件,与A1cf突变肝脏中鉴定的剪接事件存在显著重叠(图S3D)(小编注:图 S3C 的 rMAPS 基序图主要展示了在 HNF1A 缺陷细胞中,发生差异剪接的互斥外显子(MXE)周围序列里是否富集特定 RNA 结合蛋白(RBP)的识别位点。这里重点关注的是 A1CF 的识别基序。分析方法:(1)研究人员使用 rMAPS (RNA Map Analysis and Plotting Server) 工具,将复式多变量转录本剪接分析(rMATS)方法输出的差异剪接事件作为输入。(2)rMAPS 会沿着目标外显子及其上下游一定范围(通常±250 bp 或 ±500 bp)的内含子序列扫描,统计 RNA 结合蛋白(这里是 A1CF)的识别基序富集情况。(3)结果以曲线图形式展示:横坐标是相对位置(外显子边界±bp),纵坐标是基序出现的富集程度(observed/expected ratio 或 Z-score)。曲线实线表示基序分数,而虚线表示-log10 P值。上调外显子为红色曲线,下调外显子为蓝色曲线,黑色为背景基线。在 HNF1A KO 细胞的差异性 MXE 事件两侧,可以看到 A1CF 识别基序在下游约200 bp 区域显著富集(下文结果5中解释图S4B时提到A1CF识别基序在差异剪接的互斥外显子下游约200碱基对处显著富集)。曲线在这个位置出现峰值(高于背景基线),说明 A1CF 很可能结合这些互斥外显子的下游序列,从而影响它们的剪接选择。生物学意义:rMAPS 富集图把“剪接事件”与“RNA 结合蛋白作用位点”联系起来,提供了直接证据支持 A1CF 作为 HNF1A 下游效应分子,参与调控 β 细胞的可变剪接程序。图S3D:在小鼠 Hnf1a⁻/⁻ 胰岛里观察到的差异剪接事件,与 A1cf 突变肝脏中的异常剪接事件有显著重叠。这表明 A1CF 在不同组织的功能一致:缺失 A1CF 会导致类似的剪接模式异常,从而进一步证明 HNF1A 的一部分功能是通过 A1CF 间接实现的)。
这些发现共同揭示了一个先前未被认识的HNF1A功能——它通过其直接靶基因A1CF,进而调控小鼠胰岛和人类β细胞中的选择性剪接过程。
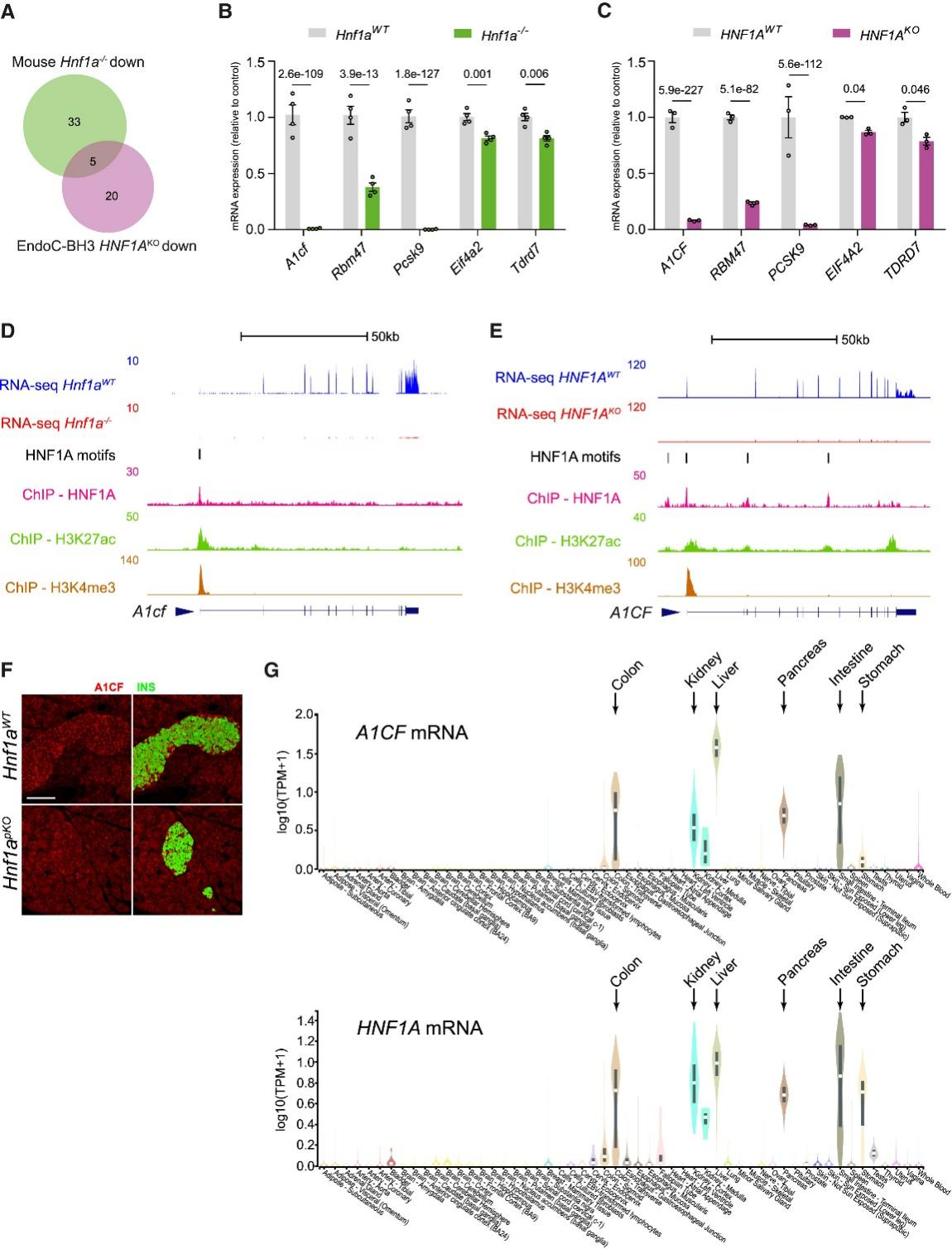
图3.HNF1A直接调节A1CF
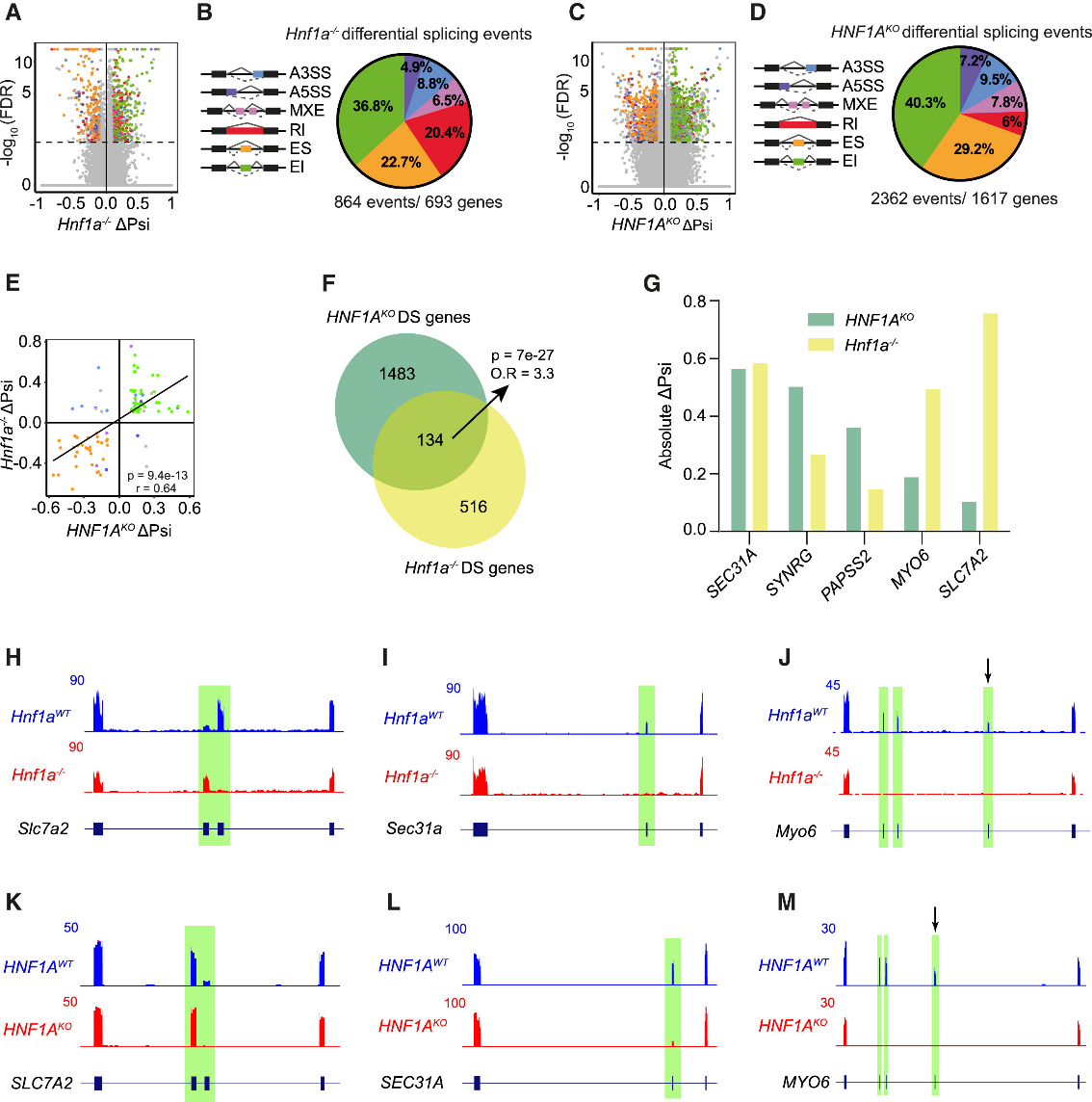
图4.HNF1A调节小鼠和人β细胞中的AS(等位基因特异性)
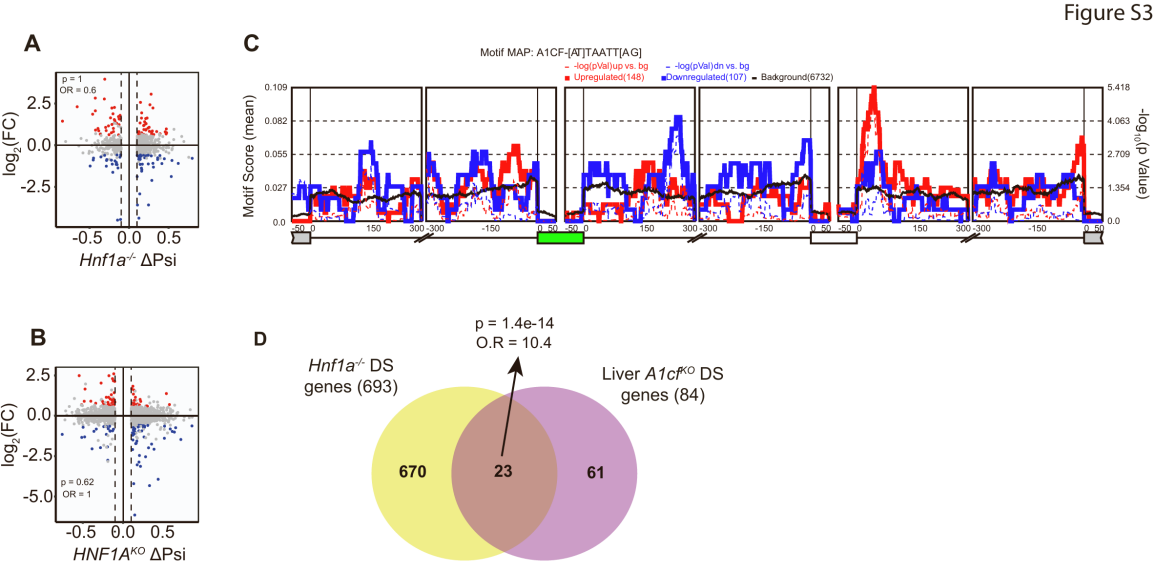
图S3.HNF1A依赖性RNA剪接分析
5、A1CF调控广泛的β细胞剪接程序,在HNF1A缺陷状态下该程序会发生紊乱
为进一步评估A1CF是否在HNF1A介导的剪接过程中发挥重要作用,研究人员通过构建纯合单外显子缺失突变体(小编注:一般KO(knockout):通常指整个基因的全长敲除或功能丧失,导致蛋白完全缺失。纯合单外显子缺失突变体:这里研究人员在 A1CF 基因里删除一个关键外显子(两条等位基因均缺失,即纯合),通过移码或缺失来破坏基因功能。这种方式更精确,可以确保敲除效果,同时避免可能的基因重排或上下游基因调控被干扰。在这项研究里,作者构建了 A1CF 纯合单外显子缺失的人胚胎干细胞(hESC) H1 系,再诱导分化为类胰岛细胞,得到的表型与常规 KO 一致(A1CF 缺失 → 剪接异常),但实验上更可控。具体构建方法为:1.gRNA设计:选择靶向 A1CF 外显子11 的序列。这个位置一旦删除,会导致移码或提前终止,从而破坏基因功能。2.组装RNP复合物:合成 crRNA 与 tracrRNA,与 Cas9 蛋白结合形成 CRISPR-Cas9 RNP。3.电转导入H1干细胞:用电穿孔将 RNP 送入 1×10⁶ H1 干细胞。4.单克隆筛选:培养、单细胞分选成克隆,用 PCR 检测纯合外显子缺失。5.建立KO细胞系:获得A1CF KO 克隆细胞,以及非靶向对照与亲本细胞。6.分化为胰岛样细胞 (SC-islets):将 KO 与对照细胞分化为胰岛样细胞,用于功能研究)成功获得A1CF敲除的EndoC-βH3细胞系。A1CFKO细胞表现出显著的选择性剪接变化,共在1,372个基因中检测到1,907个差异剪接事件(图5A、5B),其中大多数基因的表达量并无显著差异(图S4A)。与在HNF1AKO细胞中观察到的现象一致,大多数差异剪接事件为外显子跳跃或包含事件(图5B和5C),且A1CF识别基序在差异剪接的互斥外显子下游约200碱基对处显著富集(图S4B)。(小编注:“互斥外显子”指的是在pre-mRNA选择性剪接过程中,多个外显子只能有一个被保留到最终成熟的 mRNA中,它们彼此之间是“互相排斥”的关系。)
A1CF依赖性β细胞剪接程序显著富集于细胞内蛋白质靶向输送与降解过程的相关基因,功能包括:高尔基体反式网络、COPII介导的囊泡运输、囊泡靶向定位、网格蛋白介导的内吞作用、Rho GTP酶、内质网或膜运输,以及与纤毛、DNA修复和RNA剪接相关的其他功能(图5D)。由此可见,A1CF调控着一个独特的β细胞剪接程序。
A1CF与HNF1A突变体的剪接表型高度相似。在A1CFKO细胞中发生剪接改变的基因,有45%同样在HNF1AKO细胞中呈现显著剪接异常;而在HNF1AKO细胞中差异剪接的基因,有38%在A1CFKO细胞中显示异常剪接,若以p < 0.05为标准,这一比例可达59%(图5E)。关键的是,HNF1AKO细胞中的差异剪接事件在A1CFKO细胞中表现出高度相关的ΔPsi值,这表明大多数HNF1A调控的剪接异常同样也被A1CF所调控(图5F)。
在HNF1AKO和A1CFKO细胞中显示差异剪接的基因,在功能上存在共同富集(图S4D)。值得注意的是,许多β细胞功能基因在两种敲除细胞中均发生差异剪接。例如,SLC7A2基因在第7和第8外显子处呈现差异互斥剪接,且在HNF1AKO和A1CFKO细胞中均显示第8外显子包含下降(图5C、5G-5I)。在A1CFKOβ细胞中重新表达A1CF可增强SLC7A2第8外显子的表达,证实了A1CF是直接效应因子(图5J、S4E)。同样,分泌通路基因MYO6和SEC31A(图5K)以及两种突变体中众多其他异常剪接事件(小编注:MYO6(myosin VI)是肌球蛋白超家族(myosin superfamily)中 VI 类(Class VI)的“非传统肌球蛋白”,定位于高尔基/核周区,通过与 optineurin/Rab8 等形成复合体,维持高尔基体结构并促进后高尔基体—质膜方向的囊泡转运与外排(Sahlender et al. Optineurin links myosin VI to the Golgi complex and is involved in Golgi organization and exocytosis. J Cell Biol. 2005, 169(2):285-95.);其 small-insert 异构体还能将分泌颗粒系泊于皮质肌动蛋白来维持持续性外排。在胰岛β细胞中,MYO6参与了分泌小泡的内吞与囊泡运输,调节囊泡定位和膜转运。在胰岛β细胞内,抑制MYO6活性可增强胰岛素释放;机制为 MYO6与β-arrestin 协同调节GIPR内吞/去敏化与下游信号(cAMP↓、pERK↑的时空转换)(Patel NM, Sivaramakrishnan S. Myosin VI and β-arrestin synergistically regulate GIPR internalization and signaling. bioRxiv [Preprint]. 2025 Aug 28.),均发生在之前发现的具有β细胞调控功能的基因上调,包括SIDT2、SIRT6、ERC1(ELKS)、MARK2、ELAVL4(HuD)、ACSL4、ARHGEF7(βPix)和ABCC8(SUR1)等。
因此,A1CF调控着一个广泛的β细胞剪接程序,该程序涵盖众多已知的β细胞功能调节因子。这一剪接程序在HNF1A缺失细胞中严重受损,表现为A1CF表达水平及其依赖性剪接的显著下降。这表明HNF1A至少部分通过直接调控A1CF来实现对β细胞剪接过程的控制。
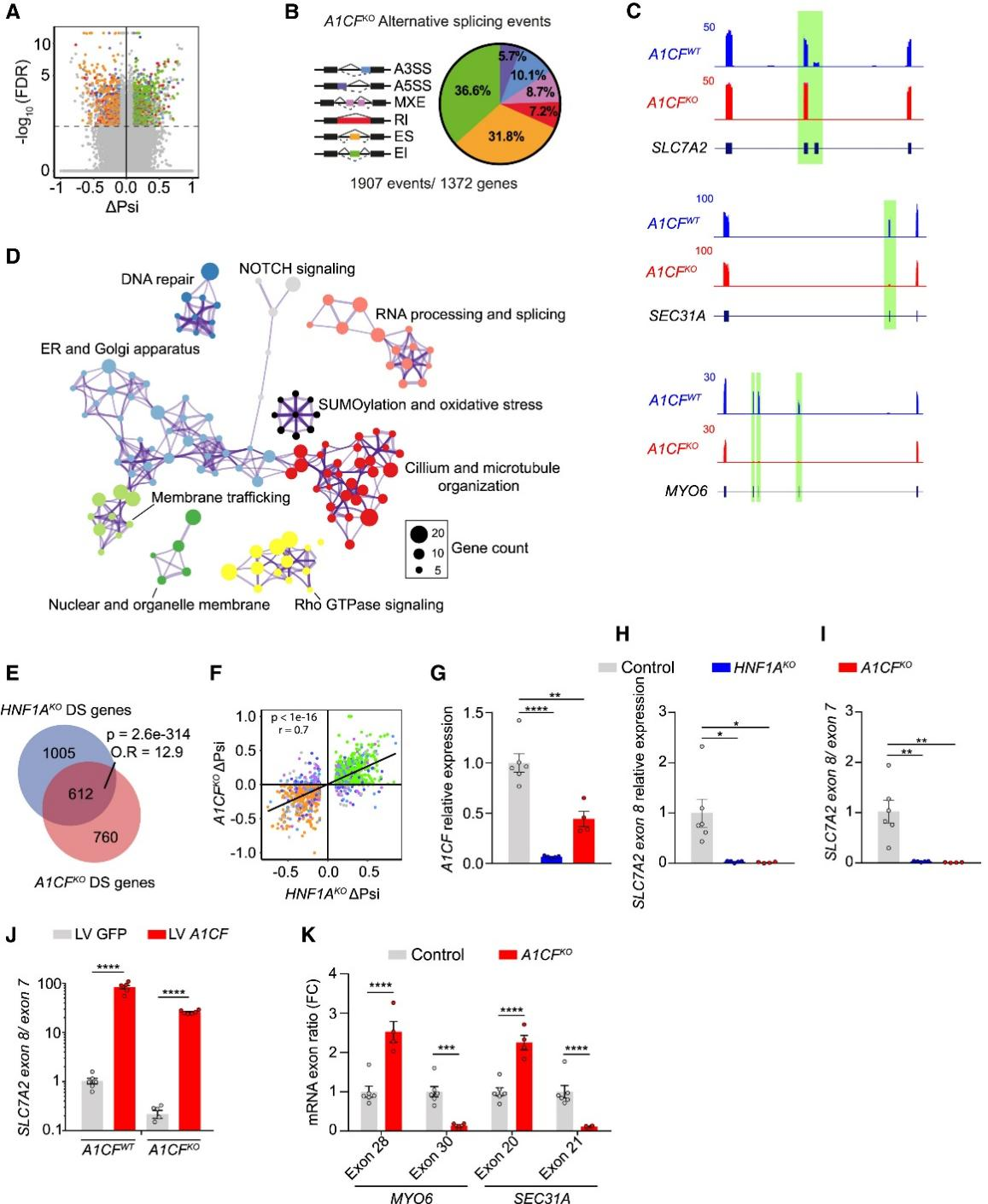
图5.A1CF敲除重现HNF1A依赖性剪接表型
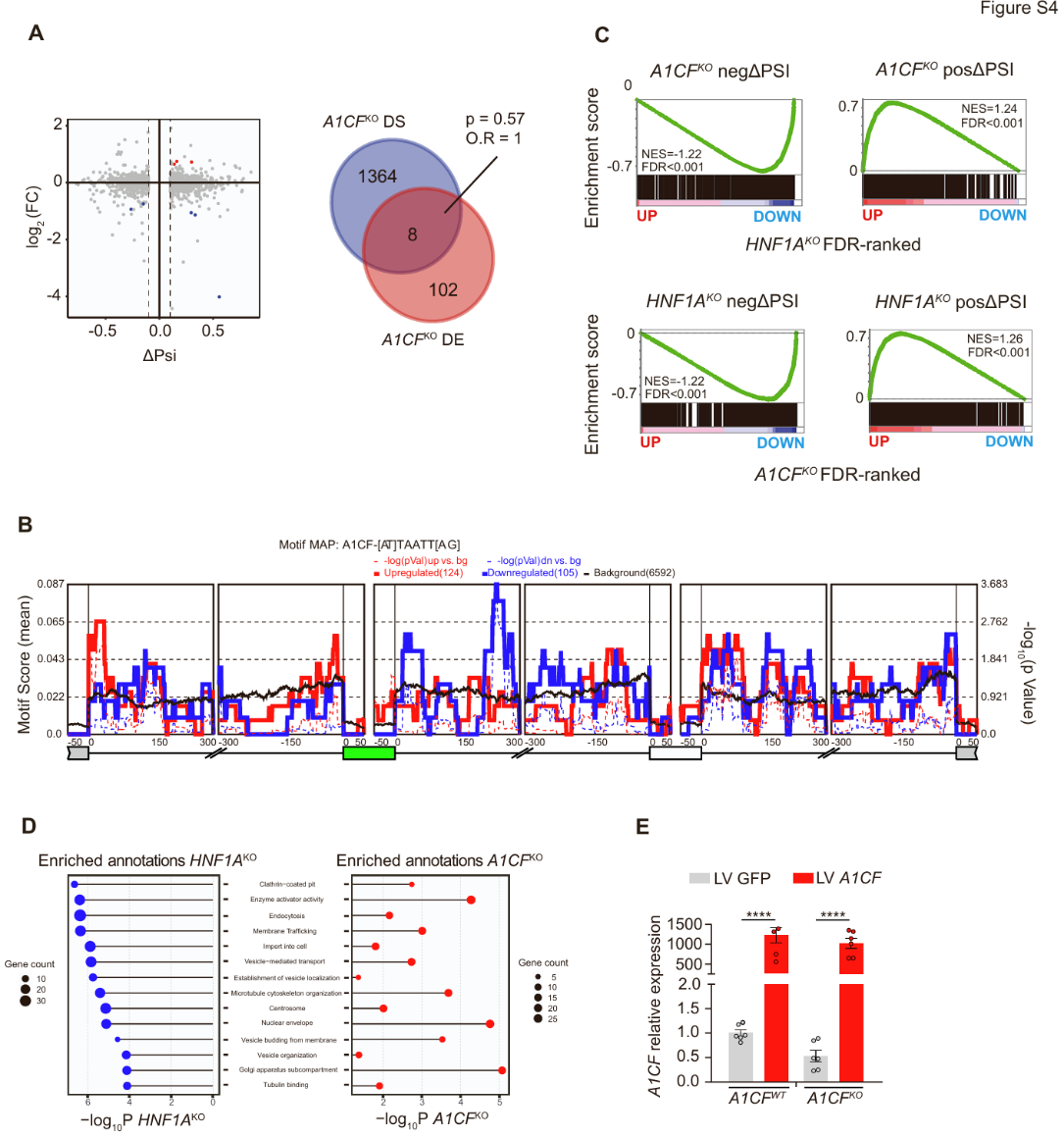
图S4. EndoC-βH3细胞A1CF与HNF1A敲除模型分析
6、A1CF 缺乏导致胰岛素分泌受损
为探究HNF1A与A1CF依赖性剪接对β细胞功能的影响,研究人员重点关注由SLC7A2基因编码的CAT2阳离子氨基酸转运蛋白。该转运蛋白可介导精氨酸(一种强效的促胰岛素分泌分子)的跨膜运输。研究模型中存在的差异性剪接SLC7A2亚型先前已被鉴定为CAT-2A和CAT-2B,其中CAT-2A包含第8号外显子(即图中SLC7A2A亚型),而CAT-2B(即图中SLC7A2B亚型)则包含第7号外显子(图6A)。CAT-2A(其在HNF1A和A1CF突变体中表达降低)亚型比CAT-2B亚型的精氨酸转运能力高出约3倍(图6A),现有研究表明这种结构特征可能介导精氨酸诱导的胰岛素分泌。
为研究SLC7A2剪接对EndoC-βH3细胞精氨酸响应的影响,研究人员采用反义寡核苷酸(ASOs)抑制A1CF mRNA的表达,或使用剪接转换寡核苷酸(SSOs)直接阻断SLC7A2第7或第8外显子的包含(图S5A–S5F)。抑制A1CF mRNA(该处理会抑制SLC7A2第8外显子的包含)以及直接阻断第8外显子的包含,均会抑制细胞对10 mM精氨酸刺激的胰岛素分泌(图S5G与S5H)。同样地,A1CFKO的EndoC-βH3细胞也表现出精氨酸依赖性胰岛素刺激作用的减弱(图6B与6C)。
研究人员进一步在人类胚胎干细胞(hESCs)中构建了A1CF外显子纯合缺失突变体,并将其诱导分化为具有葡萄糖响应性的干细胞衍生胰岛(SC islets)。该模型表现为SLC7A2第8外显子包含减少(图6D-6F)以及MYO6和SEC31A基因剪接异常(图S5I)。A1CFKO的SC胰岛表现出正常的胰岛素含量和高葡萄糖刺激下的胰岛素分泌响应,但其对10 mM精氨酸的响应性显著减弱(图6G、6H及S5J-S5M)。因此,A1CF缺陷及SLC7A2剪接改变会导致β细胞功能受损,并引发精氨酸响应异常。
鉴于HNF1A调控A1CF表达及SLC7A2剪接,研究人员接下来检测了HNF1A缺陷型β细胞的精氨酸响应。通过构建纯合HNF1AKO的干细胞衍生胰岛(SC islets)(图6I和S5N),发现与对照组相比,HNF1A缺陷模型出现显著的A1CF基因表达下调、SLC7A2第8/7外显子比率降低90%(图6J和6K),以及MYO6和SEC31A剪接异常(图S5O)。HNF1AKO的SC胰岛如预期所示表现出对葡萄糖和exendin-4的分泌响应严重受损,同时伴有β样细胞减少和胰岛素含量降低(图6I、S5P和S5Q)(小编注:Exendin-4,一种39个氨基酸的肽,是一种在毒蜥的唾液中发现的激素,是胰高血糖素样肽 1 (GLP-1) 受体的有效肽激动剂,主要通过激活胰腺β细胞上的GLP-1受体来刺激胰岛素分泌降低血糖,其人工合成品名为艾塞那肽)。这种严重的β细胞功能障碍可能由多种HNF1A调控的分子缺陷共同导致。研究人员随后在HNF1AKO SC胰岛中重新表达HNF1A,发现这能够提高A1CF mRNA水平、增加SLC7A2第8外显子包含,并增强细胞对10 mM精氨酸的胰岛素响应(图6L-6N)。由此可见,恢复HNF1A表达能增强HNF1AKO SC胰岛中的SLC7A2剪接效率和精氨酸诱导的胰岛素分泌。
研究人员发现Hnf1a突变小鼠也同样表现出Slc7a2基因剪接异常(图4H)。从4周龄Hnf1aβKO小鼠中分离出的胰岛虽仍保持正常的葡萄糖刺激胰岛素分泌功能,但其精氨酸刺激指数显著降低(图6O和6P)。在8周龄后特异性敲除β细胞Hnf1a的Hnf1aiPancKO小鼠中(图S5R),虽未出现空腹高血糖或明显的葡萄糖耐量及胰岛素分泌变化(图S5S-S5U),但在腹腔注射精氨酸后表现出血糖响应受损(15分钟血糖下降8% vs. 对照组下降25%),且胰岛素水平无显著变化(图6Q-6S)。此外,从非糖尿病Hnf1aiPancKO小鼠中分离的胰岛表现为精氨酸依赖电活动异常。尽管100 μM甲苯磺丁脲能同等程度地诱发突变组与对照组β细胞去极化(图6T和6U),但10 mM L-精氨酸仅能诱发20%的Hnf1aiPancKO β细胞产生动作电位放电和去极化(图6T-6V)(小编注:甲苯磺丁脲是一种磺酰脲类药物,它能直接刺激胰腺β细胞释放胰岛素)。综上所述,这些实验表明在小鼠和人类的HNF1A突变均能抑制精氨酸诱导的胰岛素分泌(图6A)。研究结果证实剪接因子A1CF调控β细胞功能,并揭示了HNF1A缺陷型分泌表型的潜在分子介质。(小编注:精氨酸是胰岛素强效促分泌因子,其机制是:通过阳离子转运体(如 SLC7A2/CAT2A)进入β细胞,参与膜去极化,诱导钙流入 →胰岛素释放。转运障碍会削弱 Arg-诱导的激素分泌。(Spears E, et al. Pancreatic islet α cell function and proliferation requires the arginine transporter SLC7A2. bioRxiv [Preprint]. 2023 Aug 14)。精氨酸试验(AIRarg/AIRmax)是评估β细胞分泌储备与最大分泌能力的经典方法:在高糖钳夹(≥230–450 mg/dL)下给予精氨酸可诱发最大胰岛素释放。人类疾病对应:2型糖尿病(T2D):患者胰岛β细胞常表现为精氨酸刺激下的胰岛素分泌缺陷,尤其在葡萄糖不敏感或早期β细胞功能受损时更明显。(Shankar SS, et al. Foundation for the National Institutes of Health β-Cell Project Team. Standardized Mixed-Meal Tolerance and Arginine Stimulation Tests Provide Reproducible and Complementary Measures of β-Cell Function: Results From the Foundation for the National Institutes of Health Biomarkers Consortium Investigative Series. Diabetes Care. 2016)长期 T1D/移植/残余功能:Arg 刺激可检出残余分泌,即使对葡萄糖无反应(Marc Gregory Yu, et al. Residual β cell function and monogenic variants in long-duration type 1 diabetes patients. JCI 2019)。因此,目前研究显示:精氨酸可作为一种补充机制参与胰岛素相关的代谢调控,为糖尿病的防治提供额外的调控策略。)
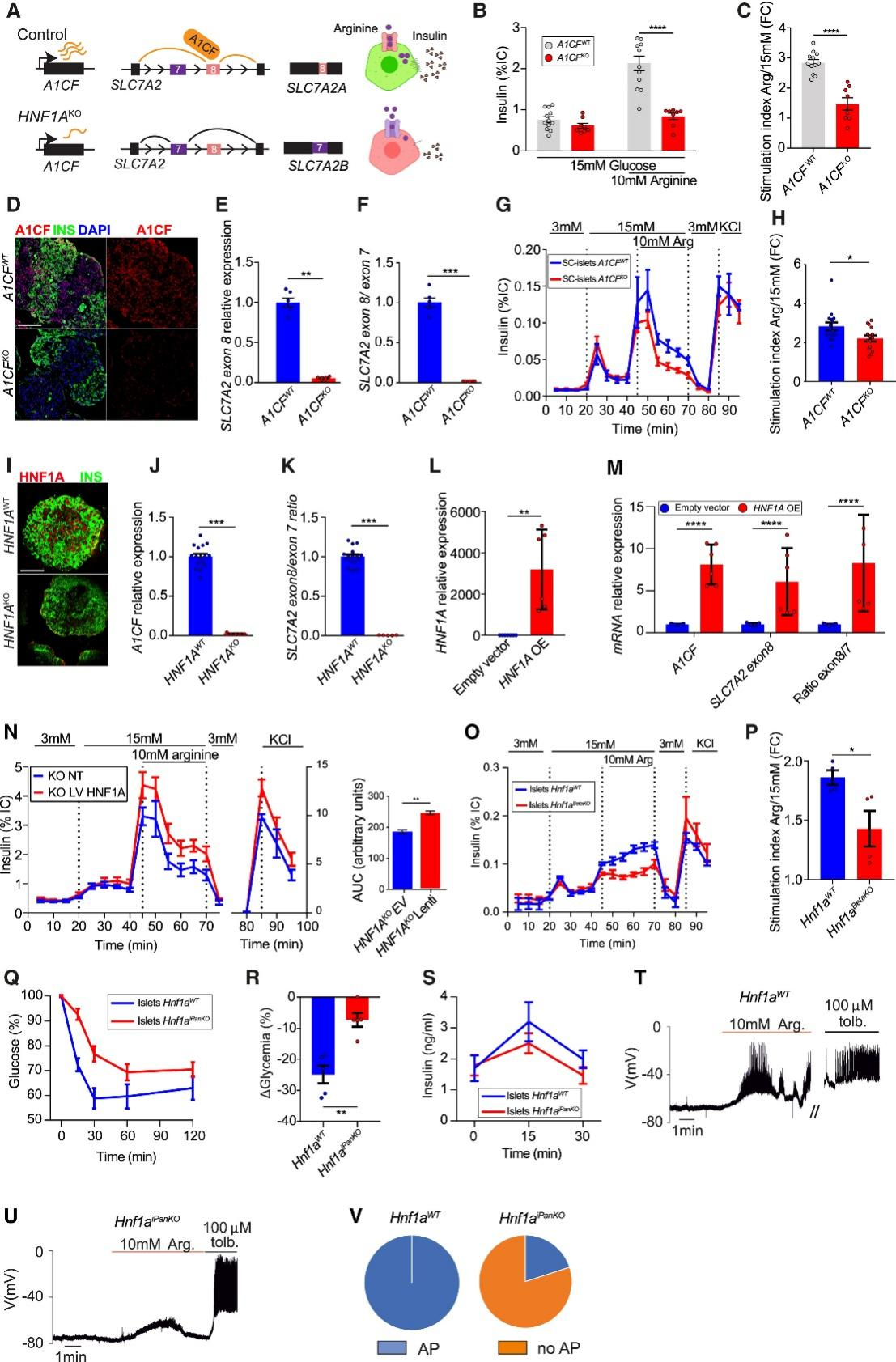
图6.异常的A1CF依赖性剪接导致胰岛素分泌受损
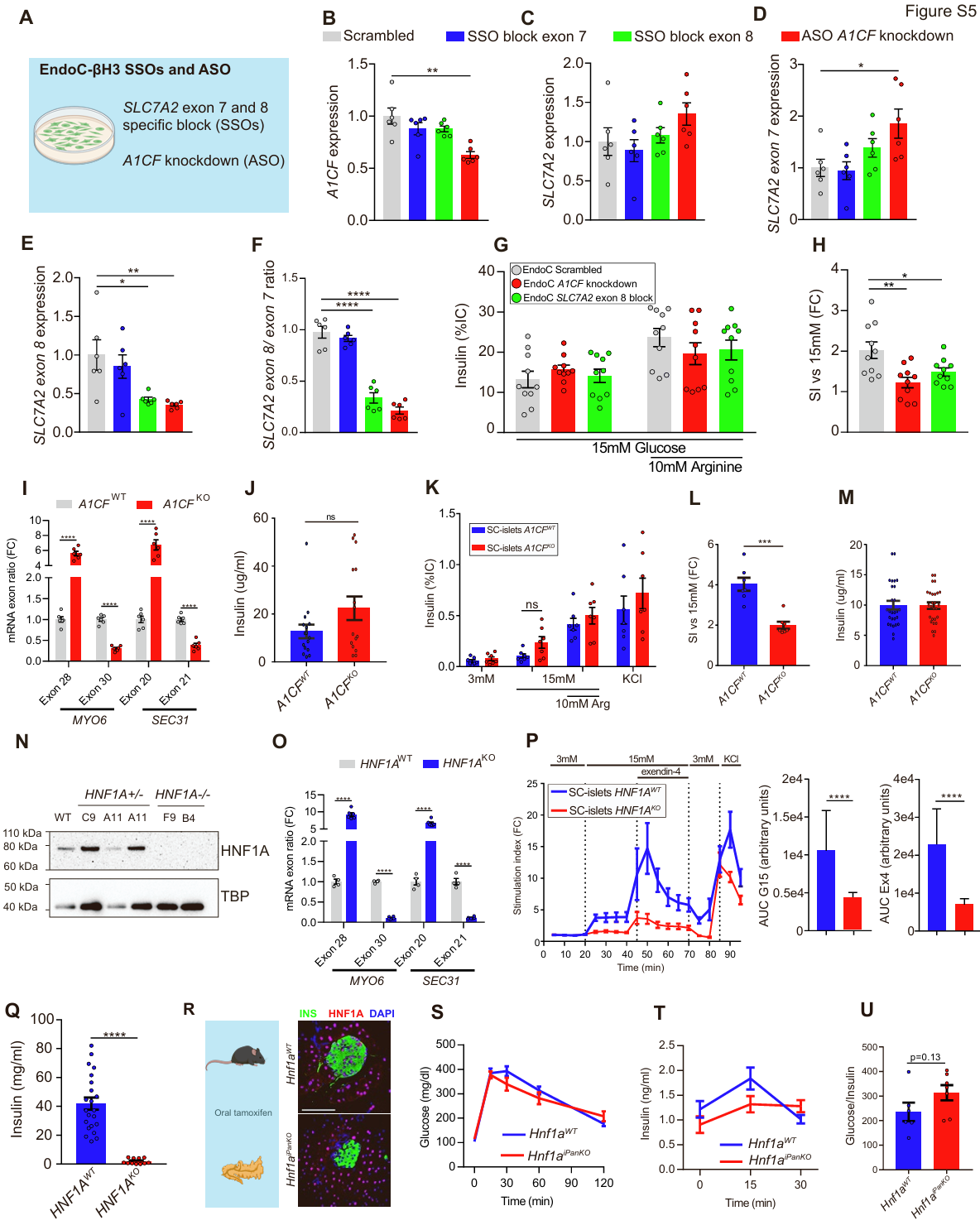
图S5.A1CF依赖性剪接和β细胞功能
7、T2D相关β细胞亚群中A1CF依赖性剪接变化
研究人员接下来试图寻找HNF1A影响人类原代β细胞中A1CF依赖性剪接的证据。近期研究表明人类β细胞具有异质性,其转录状态因HNF1A活性而异。研究人员推测,若HNF1A在天然人类胰岛中调控A1CF表达,则HNF1A活性较高的β细胞应表现出更强的A1CF依赖性剪接。为此,研究人员分析了来自人类细胞图谱ESPACE联盟的单细胞转录组数据(通过VASA-seq技术生成)——该技术因能捕获单细胞全长转录本而适用于RNA剪接分析。利用来自11个个体(5例T2D患者,6例非糖尿病患者)的人类胰岛样本合并VASA-seq数据,研究人员首先对主要细胞类型进行聚类,获得2,036个β细胞(图S6A),并定义了主要β细胞簇(图7A)。随后研究人员计算了每个β细胞簇中172个HNF1A靶基因(与HNF1A结合且在HNF1AKO细胞中下调的基因)的表达量,结果发现β2细胞簇具有显著更高的HNF1A活性(图7B)。这些HNF1A高活性β细胞相对于其他所有β细胞表现出广泛的差异性剪接(图S6B),且这些剪接变化在A1CFKOβ细胞的异常剪接事件中显著富集(图7C和S6C)。因此,β细胞状态不仅因HNF1A活性而异,还表现出A1CF依赖性剪接的差异。这些结果与HNF1A在天然人类β细胞中A1CF依赖性剪接调控机制相一致。
为评估这一发现对2型糖尿病(T2D)的意义,研究人员重点关注先前与T2D相关的β细胞状态。近期一项研究通过机器学习区分出β1和β2细胞状态:这两种状态在T2D供体的胰岛中分别呈现缺失或富集现象,且相应地表现出HNF1A活性升高或降低的特征。通过分析数据中的β1和β2基因标记集,发现HNF1A高活性β细胞具有独特的β1基因特征,另外两个细胞簇呈现显著β2特征,而剩余细胞则低表达这两种基因集(图7A和7B)。在T2D个体供体中,HNF1A活性较低的β2细胞增多,而β1(HNF1A高活性)细胞减少(图7D),且T2D供体的β2/β1细胞比率较非T2D供体高约8倍(图7E)。因此,这些数据证实T2D的发生会伴随着β1和β2细胞状态的明显转变。
研究人员随后探究了与T2D相关的β细胞状态中的剪接情况,发现A1CFKO细胞中内含子保留率降低的剪接事件在β2细胞中同样呈现下降趋势,而A1CFKO细胞中内含子保留率升高的事件在β2细胞中较β1细胞上升。具体而言,在A1CFKO细胞中呈现负调控的剪接事件,其β2与β1细胞的ΔPsi值为-0.192;而呈现正调控的剪接事件对应的值为0.071(图7F)。例如介导精氨酸转运的SLC7A2外显子8异构体——该异构体在HNF1AKO和A1CFKO细胞中表达降低——在T2D相关的β2细胞中也被抑制(图7G)。同样地,与T2D相关的β2细胞重现了HNF1AKO和A1CFKO细胞在SIRT6和SEC31A基因上的差异剪接特征(图S6D)。这些结果表明,T2D中的β细胞状态改变不仅涉及基因表达差异,还伴随着A1CF依赖性RNA剪接的异常调控。
8、A1CF 的变化显示与T2D和血糖特征有关
为深入探究A1CF在人体葡萄糖稳态中的作用,研究人员通过近期大规模荟萃分析测评了A1CF基因的遗传变异是否影响血糖水平和2型糖尿病(T2D)风险。研究显示,A1CF基因变异与随机血糖水平及T2D风险显著相关(图7H)。经过对遗传信号的精准定位后发现,位于人类胰腺胰岛增强子区域的候选致病突变体,通过染色质互作图谱与A1CF启动子相连接(图7I)。尽管它们的空间位置邻近,基于较低的连锁不平衡度(LD)和较弱的遗传共定位证据来看,影响血糖与T2D风险的关联信号可能具有遗传独立性(小编注:连锁不平衡是指在某一群体中,分属两个或多个基因座的等位基因同时出现在一条染色体上的几率高于随机出现的频率。这意味着这些等位基因并不是独立遗传的,而是以某种方式相互关联)。为验证A1CF是否为这些关联的潜在效应基因,研究人员在约400例人类胰岛RNA测序样本中进行了全转录组关联分析(TWAS),发现胰岛A1CF表达与随机血糖存在显著关联(图S6E)。相应地,A1CF变异位点rs61856594可调控人类胰岛A1CF的eQTL,并与随机血糖关联呈现遗传共定位(图7J、7K)(小编注:eQTL是指基因组上的一个基因座,可以理解为它是一个“基因表达开关”)。T2D变异位点rs12570156与多个数据集中的胰岛和整个胰腺组织A1CF的eQTL也显著性关联(图7L)(小编注:这里图7L只给出了全胰腺组织的eQTL,胰岛的数据集是引用自其他文献)。
对随机血糖相关位点(rs61856594)和T2D相关位点(rs12570156)的深入分析表明,那些能提高A1CF基因表达水平的等位基因会导致更低的随机血糖水平;降低T2D患病风险;改善β细胞功能、葡萄糖耐量和急性胰岛素反应(BIGTT-AIR);并与较高的出生体重及较大的胰腺体积相关——孟德尔随机化和多基因研究均已证实这两种性状与胰岛素分泌功能密切相关(图7M)。与A1CF在其他组织中的表达模式一致,这些相同等位基因还具有多效性作用,特别是能显著升高肝脏中γ-谷氨酰转移酶的水平(图7M),而一个低频的A1CF错义变异则与高甘油三酯血症相关。由此可见,与功能研究得出的预测相一致,人类遗传学证据支持以下结论:胰岛A1CF表达水平的提高可增强β细胞功能、改善葡萄糖稳态并降低T2D易感性。
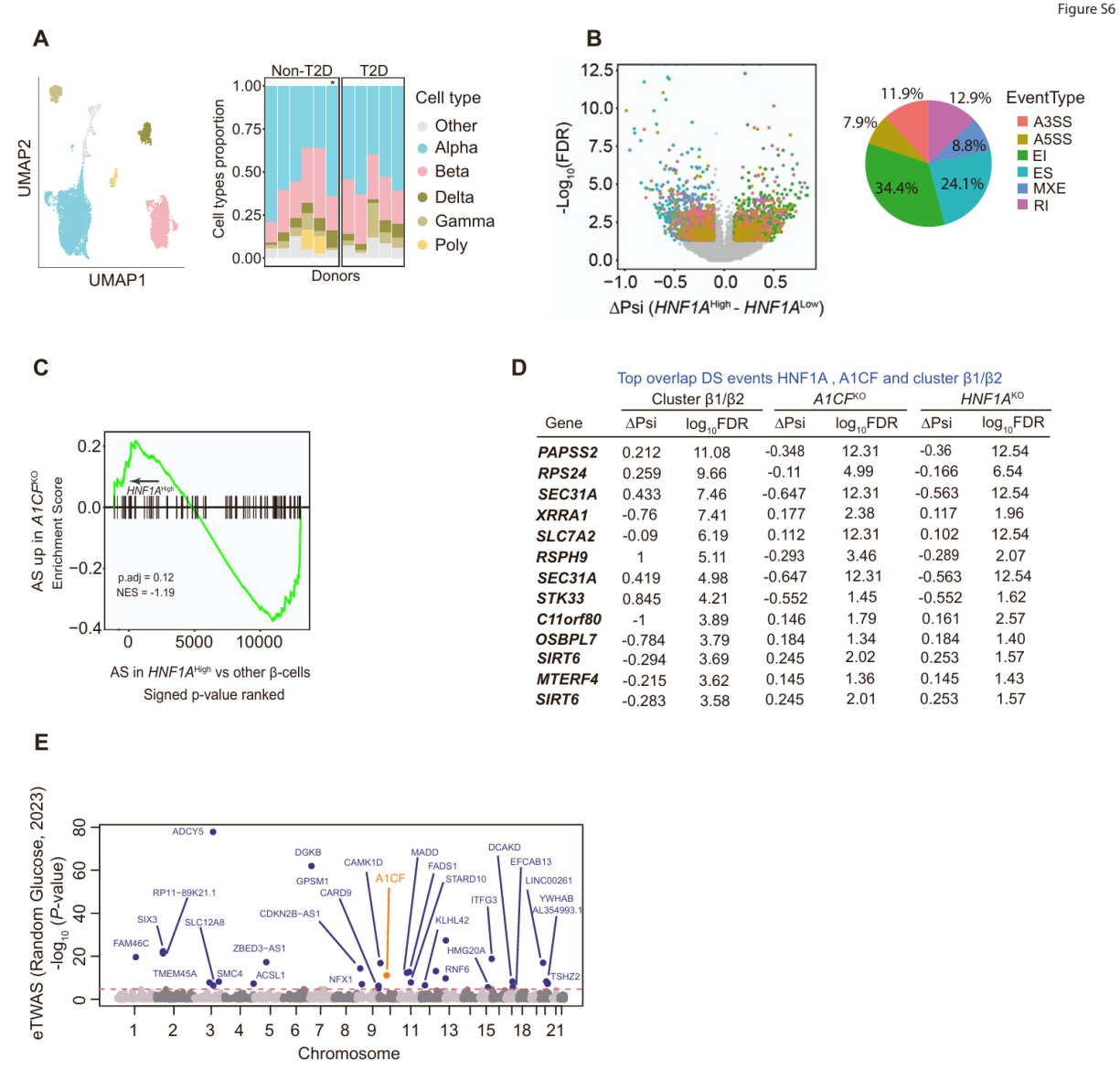
图S6. A1CF表达在T2D中的遗传学分析
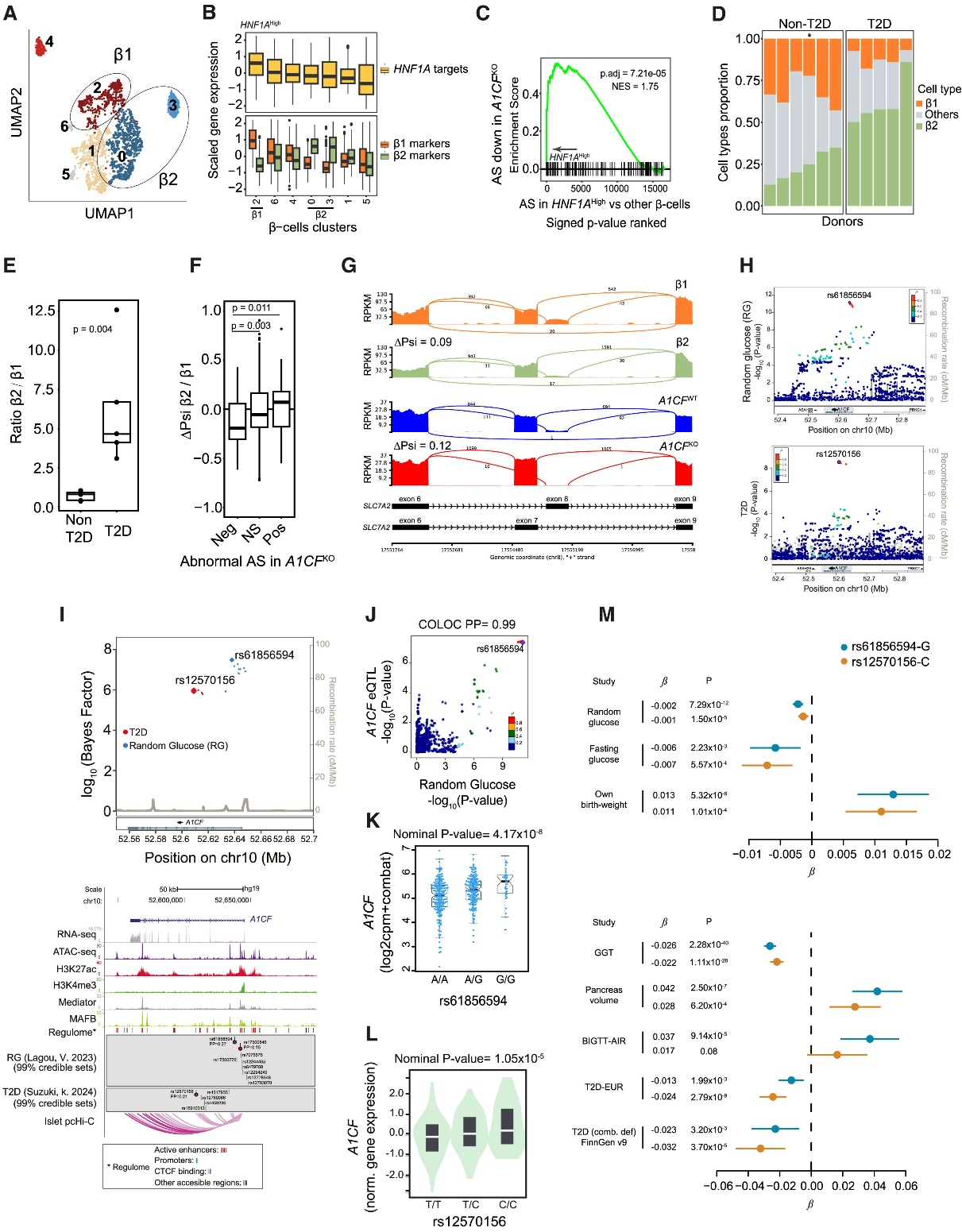
图7.β细胞和T2D中的A1CF依赖性剪接
总结
本研究证明,HNF1A通过直接激活A1CF,在β细胞内形成一条关键的转录-剪接调控轴,该机制对维持β细胞正常功能至关重要。研究进一步证实,A1CF介导的剪接异常是HNF1A缺陷型单基因糖尿病和T2D的分子病理基础。这一发现不仅为HNF1A缺陷型糖尿病的治疗提供了以RNA剪接为靶点的新策略,也为深入理解糖尿病发病机制中转录调控-剪接轴提供了重要依据。
原文链接:https://www.sciencedirect.com/science/article/pii/S1550413125003341?via%3Dihub
https://blog.sciencenet.cn/blog-3483272-1506654.html
上一篇:Cell metabolism:脂肪罢工,神经酰胺是元凶
下一篇:代谢学人——Science:巨噬细胞的双重人生