博文
快速构建高质量系统发生基因组学数据集:脚本使用和指南
||
近日,南京农业大学植物保护学院昆虫学系张峰团队在期刊《Zoological Systematics》在线发表了题为“Construction of a phylogenetic matrix: Scripts and guidelines for phylogenomics”的论文。该研究构建了一套自定义脚本,用于系统发生基因组学中通用单拷贝直同源基因(universal single-copy orthologs;USCOs)的提取、序列比对、位点剪切、标记过滤和数据集(matrix)创建等分析,旨在构建系统发生树之前简化分析流程并提高树的准确性。这些脚本采用了一系列计算高效的生物信息学工具,并通过LINUX系统中类似Windows界面的“拖拉”操作,与通用的‘BASH’命令或可视化界面一起使用。
相关脚本下载和使用说明详见:https://github.com/xtmtd/Phylogenomics/tree/main/scripts。此外,我们还提供了整合所有分析软件、脚本及测试数据的虚拟机镜像(账号:zf;密码:1):https://dx.doi.org/10.6084/m9.figshare.21283026;以及每个脚本的操作短视频:https://space.bilibili.com/319699648/channel/seriesdetail?sid=2682055,供大家学习参考。
由于近年来高通量测序技术的快速发展、各类群基因组数据的不断丰富,利用系统发生基因组学分析手段解决物种间的进化关系已经成为了主流。然而,仅仅增加分子标记数量并不能有效地解决某些关键节点物种间的亲缘关系。随着数据量的增加,系统性误差(又叫分析误差)也随之增加。数据缺失(missing data)、组成异质性(compositional heterogeneity)、速率异质性(rate heterogeneity)和不完全谱系分选(incomplete lineage sorting)是目前常见的几类系统性误差之一,对构建准确的系统发生树产生了较大影响。因此,想要降低这些误差带来的影响,就需要创建高质量的、可靠的matrix,从而构建准确的、稳定的系统发育树。因此,我们开发了一套适用于大多数动物类群(已在弹尾纲、半翅目、膜翅目、鳞翅目等类群成功测试)的系统发生基因组学分析脚本以及相应的分析流程,旨在快速构建高质量的matrix,从而提高系统基因组树的准确性。
文中提供的脚本和流程适用于多种分子标记,如USCO,UCE,AHE等。构建高质量matrix的过程主要分为5个步骤,每一步即是独立的又是相互联系的,大家可以根据需要选择合适的步骤进行分析:
步骤1,USCO分子标记提取,即从BUSCO产生的文件夹(run_*)中提取氨基酸和核苷酸序列。依据基因名,将所有物种拥有相同基因的序列合并在一个FASTA文件中,并过滤物种数小于3的基因标记(使用的脚本:BUSCO_extraction.sh)。
步骤2,序列比对。可以自行选择MAFFT或MAGUS任一比对软件,并设置合适的参数进行序列比对(使用的脚本:align_MAFFT.sh)。
步骤3,位点剪切。我们提供了三种不同的剪切工具:trimAl、BMGE或ClipKIT。可以选择任一工具,并设置合适的参数进行位点剪切(使用的脚本:trimming_alignments.sh)。
步骤4,标记过滤,依据基因属性又分为“基于序列的标记过滤”和“基于基因树的标记过滤”。脚本中整合了包括序列长度计算、简约信息位点数量统计、组成异质性检测(例如:GC含量,RCV)、同质性检测(例如:symtest)、进化速率计算、虚假同源序列鉴定、高系统发育信号识别以及“不一致”的基因(导致基因树和物种树不一致的基因)识别等十几种不同的过滤策略。根据脚本提示,选择合适的过滤策略及参数,可进行分子标记(基因)过滤(使用的脚本:loci_filtering_alignment-based.sh和loci_filtering_tree-based.sh)。
步骤5,序列合并,即创建高质量matrix。可将以上步骤完成后的分子标记,在考虑不同完整性(代表了所有分区中物种所占的最低比例,通常在50% ~ 100%之间)的情况下进行序列合并,产生“超级矩阵”(supermatrix)。此外,我们在此步骤中添加了一个额外的功能,即将外群物种放置“超级矩阵”中的首行,使其在最终的树文件中更加直观(使用的脚本:matrix_generation.sh)。
本研究突出了高质量matrix对系统发生推断的影响,推动了基于“大数据”系统发育研究的进展。为了进一步优化和提高系统发生基因组学的分析过程,我们将利用多种策略(方法和模型)进行系统发生树的构建。此外,未来的研究也将受益于越来越丰富的高质量基因组数据,从而挖掘更多潜在的基因组信息(例如:基因组重复、重排和共线性区块等)进行系统发生学的研究。相信我们开发的脚本和流程将有助于提高系统发生树的便捷性和准确性。
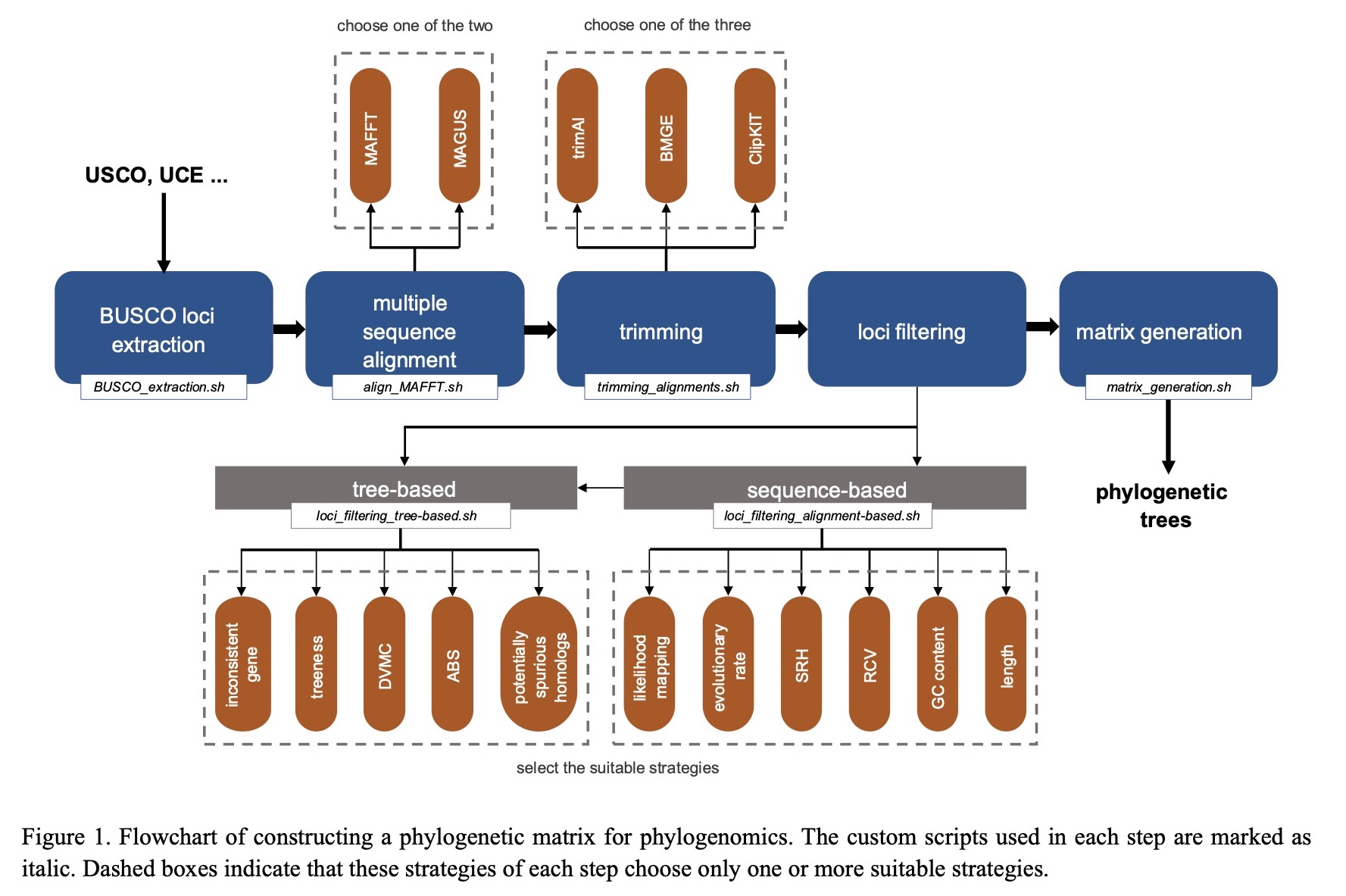
系统发生基因组学分析简化流程
本研究得到了国家自然科学基金项目(31970434,32270470)、江苏农林职业技术学院青年扶持项目(2022kj27)的资助。南京农业大学植物保护学院杜诗雨博士生为论文第一作者,南京农业大学植物保护学院张峰教授为论文通讯作者;江苏农林职业技术学院丁银环博士、中国农业大学李虎教授、首都师范大学张爱兵教授、中国科学院动物研究所罗阿蓉副研究员和朱朝东研究员参与了研究和写作。
原文链接:
https://www.zootax.com.cn/EN/10.11865/zs.2023201
或https://www.zootax.com.cn/EN/Y2023/V48/I2/107
https://blog.sciencenet.cn/blog-536560-1387553.html
上一篇:关于召开“中国昆虫学会第十八届昆虫分类区系学术研讨会”的通知 (第二轮)
下一篇:在系统发生树上整合功能性状和DNA条形码
