博文
氢水联合吸入氢气预防小鼠皮肤癌
||
氢水联合吸入氢气预防小鼠皮肤癌
氢气对癌症治疗的效应在逻辑上仍然存在模糊性,但预防癌症的发生非常符合逻辑。癌症发生的因素很复杂,但紫外线等环境和自身因素诱导的氧化损伤基因突变是其中非常重要的原因,而组织炎症反应也是导致癌症发生发展的重要微环境因素。氢气减少氧化应激和炎症反应的作用非常明确,因此预防癌症的发生也非常具有理论可行性。既往研究发现,氢水对预防原发性肝癌,预防胆管硬化和癌变,预防结肠癌,各种射线诱导的癌症。现在最新研究发现氢水联合吸入氢气预防皮肤紫外线诱导的癌症发生。给氢气预防癌症增加了新的研究证据。虽然关于人类癌症预防的证据相对比较少,但北京协和医院管教授近日学术会议上报道了氢水饮用预防食管癌的临床研究,给这一方向增加了人类临床研究证据。如果氢气疗法用于人类癌症预防的效果明确,作为一种安全可行的大众化工具,这种方法在癌症防控领域的价值将会非常突出,具有革命性意义。
以上为个人观点。
氢气减轻慢性炎症并延缓紫外线 B 诱导的小鼠皮肤癌发生
氢气(H₂)具有抗炎和抗氧化特性。然而,其在紫外线 B(UVB)诱导的皮肤癌发生中的作用仍不清楚。雄性 HR‑1 无毛小鼠在慢性背部 UVB 照射期间(270 mJ/cm²,每周 3 次,共 20 周)持续接受 H₂(2% 氢气吸入 + 富氢水 HRW)或对照处理(正常空气 + 去氢水),随后进行 10 周观察期。该实验方案独立重复一次。H₂暴露在两个实验系列中均一致地延缓了乳头状瘤的出现并减少了累积肿瘤数量,而生存时间延长和鳞状细胞癌(SCC)发生延迟仅在其中一个实验系列中达到统计学显著性。环丁烷嘧啶二聚体(CPD)水平保持不变,表明 DNA 光损伤未减少。H₂暴露降低了表皮 T 细胞浸润、真皮 IL‑6 水平以及核磷酸化 STAT3 水平。ERK 和 JNK 磷酸化水平也降低。H₂在急性 UVB 暴露后维持了 GSH/GSSG 比率,并在慢性暴露期间减少了核 Nrf2 积累。表皮厚度和增殖标志物(Ki‑67 和 PCNA)均降低。这些发现表明,持续给予 H₂通过调节 IL‑6/STAT3 和 ERK/JNK 通路减轻炎症相关的早期 UVB 致癌过程,支持其作为一种化学预防策略的应用。
1. 引言
鳞状细胞癌(SCC)是仅次于基底细胞癌的第二常见皮肤癌[1]。根据 2016–2018 年的数据,日本每年约报告 7700 例[2],而美国在 2021 年记录了约 180 万例[3],凸显了 SCC 在全球范围内的重大公共卫生负担。这种恶性肿瘤主要影响老年人群,约占所有皮肤癌的 20%[1]。慢性紫外线(UV)暴露,特别是紫外线 B(UVB,280–320 nm),是主要致病因素[4],头颈部等日光暴露部位更易受累[1]。UVB 可直接造成 DNA 损伤并产生活性氧(ROS),从而启动复杂的炎症级联反应[4]。角质形成细胞释放多种炎症介质,包括 IL‑1β、IL‑6、TNF‑α 和 IL‑8,形成促炎微环境[5],促进肿瘤发生的各个阶段——从初始增殖到转化细胞的侵袭[6]。由 IL‑6 信号激活的 JAK/STAT3 通路是炎症与致癌之间的关键分子连接[7]。STAT3 激活后会上调多个靶基因,包括抗凋亡因子 Bcl‑2、细胞周期调节因子 cyclin D1 和 VEGF,共同促进癌细胞存活和增殖[7]。与正常皮肤相比,人 SCC 和基底细胞癌标本中磷酸化 STAT3 的表达与肿瘤浸润深度呈正相关[8,9]。这些发现表明,ROS‑STAT3 信号轴是 UVB 诱导皮肤癌发生的核心分子靶点。
氢气(H₂)作为一种生物活性气体,可减少氧化应激和炎症,提示其在多种病理状态中具有治疗潜力[10,11]。H₂通过调节上游信号通路(包括激活 Nrf2、抑制 NF‑κB 和 NLRP3 炎症小体、以及抑制 STAT3 磷酸化)来减轻 ROS 生成和炎症细胞因子分泌[12]。在 UVB 照射大鼠的皮肤组织中,富氢盐水可降低炎症细胞因子和氧化应激标志物,从而减轻皮肤损伤[13]。使用 HaCaT 角质形成细胞的体外研究表明,H₂可抑制 UVB 诱导的 ROS 生成和脂质过氧化[14]。此外,H₂治疗可在多种器官和肿瘤模型中降低 STAT3 磷酸化和 Bcl‑2 表达[15,16,17]。由于 STAT3 激活在 SCC 发展中起关键作用,这些观察结果提示 H₂可能通过在早期 UVB 暴露期间阻断 ROS‑炎症‑STAT3 级联反应来预防皮肤癌发生。
本研究探讨了 H₂是否通过抑制慢性 UVB 暴露期间的炎症和 STAT3 激活来延缓 SCC 发生。我们让无毛小鼠接受长期 UVB 照射,同时通过环境暴露和饮用富氢水(HRW)持续给予 H₂。结果表明,H₂治疗显著延迟了初始病变的形成,并呈现出 SCC 发生延迟和生存延长的趋势。这些结果支持将 H₂作为一种针对皮肤癌发生中 ROS‑炎症‑STAT3 级联反应的可行预防干预手段。
2. 结果
2.1 H₂减轻短期 UVB 照射诱导的急性皮肤反应
我们在长期 H₂给药条件下观察了短期 UVB 照射后的急性光炎症反应。4 周龄小鼠被随机分为对照组或 H₂治疗组。H₂治疗组小鼠自由饮用 HRW,并饲养在含 2% 氢气的环境中。小鼠在 7 周龄时开始接受 UVB 照射(270 mJ/cm²,每周 3 次)(图 1)。照射 5 周后,两组之间出现明显的皮肤红斑差异(图 S1),提示急性光炎症损伤减轻,与先前报道一致[18]。
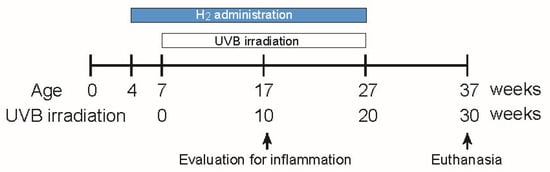
图 1. 慢性 UVB 暴露和 H₂治疗的实验示意图。4 周龄 HR‑1 无毛小鼠被随机分为 H₂组(2% 氢气吸入 + 富氢水 HRW;溶解 H₂浓度 ≥ 0.8 mM)或对照组,并从 7 周龄至 27 周龄接受背部 UVB 照射(270 mJ/cm²,每周 3 次)。在照射第 10 周评估炎症和 ROS。每周计数肿瘤,并在 37 周龄时处死小鼠进行组织学评估。
2.2 H₂延缓慢性 UVB 诱导肿瘤发生中的早期病变形成,并呈现 SCC 发生延迟和生存延长趋势
如先前报道[19],无毛小鼠在 UVB 照射后先出现乳头状瘤,随后恶变为 SCC。在本研究中,UVB 照射开始约 10 周后偶尔观察到直径约 1 mm 的小肿瘤(图 2A)。此外,在初始 UVB 暴露后 30 周,小鼠背部皮肤形成了各种大小的肿瘤(图 2B)。组织病理学检查显示,这些小肿瘤具有乳头状瘤的典型特征(图 2C),而 30 周时观察到的大多数肿瘤则表现为 SCC 的形态特征(图 2D)。
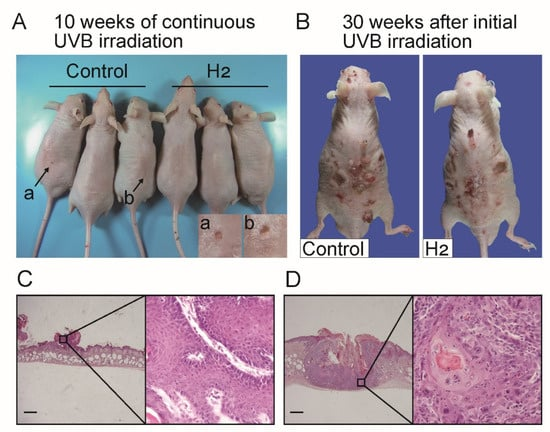
图 2. UVB 诱导肿瘤的大体和组织学表现。(A)连续 UVB 照射 10 周后的背部图像;(B)初始暴露后 30 周的背部图像。(A)中箭头指示对照组中观察到的代表性小结节;插图为标记(a,b)病变的高倍视图。与对照组相比,H₂给药减少了小结节数量,并在 30 周时呈现肿瘤数量减少的趋势。组织学检查显示,约 10 周后出现的小结节具有乳头状瘤特征(C),而 30 周时大多数病变为侵袭性 SCC(D)。比例尺 = 500 μm。
为进一步表征肿瘤进展,我们检查了 UVB 照射开始后 30 周形成的肿瘤的组织病理学特征。随机选择乳头状瘤和 SCC 并测量其直径。如图 S2 所示,直径约 1 mm 的乳头状瘤不超过 5 mm。相比之下,SCC 的直径范围为约 3–10 mm。
基于这些观察,我们每周测量肿瘤直径以研究 H₂对肿瘤增殖的影响。首次出现时直径 ≥1 mm 且 ≤2 mm 的肿瘤被归类为乳头状瘤(SCC 发展的初始病变),而直径 ≥5 mm 的肿瘤被归类为晚期 SCC。我们仔细监测了肿瘤形成的时间进程。H₂治疗组的乳头状瘤出现时间显著延迟,且总肿瘤数量显著减少(图 3A,B)。直径 ≥5 mm 的肿瘤出现时间被用作恶性进展为 SCC 的指标。尽管未观察到统计学显著差异,但 H₂治疗组呈现出明显的致癌延迟趋势(图 3C)。在生存分析中,无毛小鼠在照射后被监测 30 周,结果显示 H₂给药组的生存率显著高于对照组(图 3D)。
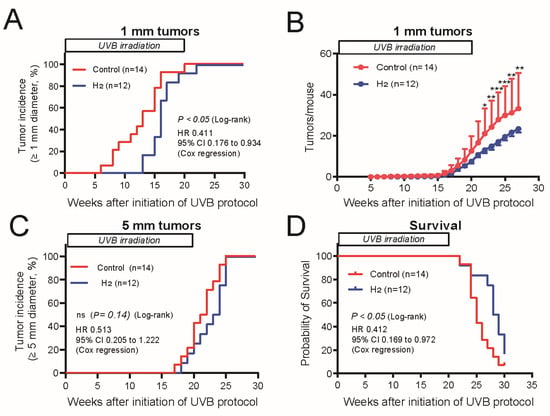
图 3. H₂延缓慢性 UVB 诱导肿瘤发生中的早期病变形成,并呈现 SCC 发生延迟和生存延长趋势。(A)对照组和 H₂组中出现肿瘤(直径 ≥1 mm)的小鼠百分比随 UVB 暴露周数的变化。(B)每组每只小鼠的总肿瘤数量随时间的变化。(C)出现肿瘤(直径 ≥5 mm)的小鼠百分比随时间的变化。(D)对照组和 H₂组的生存曲线。(A,C,D)采用 Kaplan–Meier 法估计肿瘤发生率和生存率,log‑rank 检验比较差异,Cox 比例风险回归估计风险比(HR)和 95% 置信区间(CI)。(B)采用双因素 ANOVA 结合 Šidák 事后检验分析组间差异。*p<0.05;p<0.01;*p<0.001。
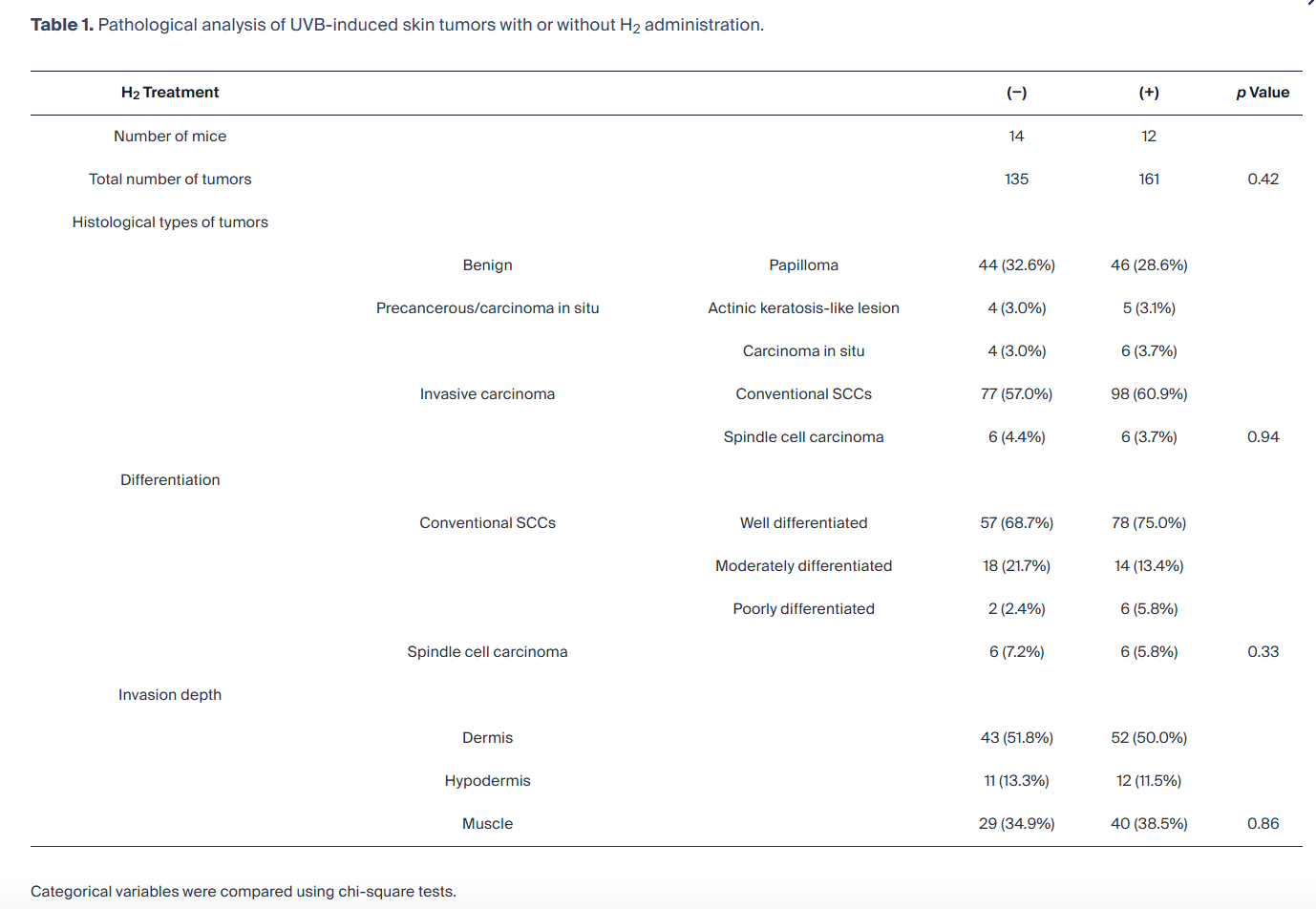
为验证 H₂对 UVB 诱导皮肤癌的抑瘤作用的可重复性,我们使用相同方案重复了实验。在该实验系列中,H₂显著延迟了乳头状瘤的出现(图 S3A),减少了乳头状瘤总数(图 S3B),并显著延迟了 SCC 的发生(图 S3C),同时呈现出生存延长的趋势(p=0.081)(图 S3D)。对 UVB 照射开始后 30 周收集的肿瘤组织进行病理检查。对照组中,28 周后肿瘤增大并融合,导致可用于病理评估的肿瘤数量减少,但差异无统计学意义。H₂给药未改变 UVB 诱导癌的组织学表现或特征(表 1)。
表 1. 有无 H₂给药时 UVB 诱导皮肤肿瘤的病理分析。
2.3 H₂不抑制 UVB 诱导的皮肤嘧啶二聚体形成
我们假设 H₂给药通过影响嘧啶二聚体形成(UVB 诱导皮肤中主要的 DNA 损伤)来影响 UVB 诱导皮肤癌的发展。因此,我们检查了在 UVB 照射前接受 H₂预处理 3 周的无毛小鼠皮肤中的 CPD 形成,并与未处理对照组进行比较(图 S4)。H₂给药组和对照组之间未观察到 CPD 形成的差异,表明 H₂的保护作用通过不依赖于预防 DNA 损伤的机制实现。
2.4 H₂减轻 UVB 诱导的皮肤炎症
H₂的一个公认生物学效应是减轻炎症[11,20]。鉴于炎症在 UVB 诱导皮肤癌进展中的关键作用[5,21],我们研究了 H₂在多大程度上减少 UVB 诱导的炎症。在 UVB 照射开始后 10 周收集无毛小鼠背部皮肤样本,并通过免疫组化染色检查炎症细胞分布(图 4A)。与对照组相比,H₂治疗的无毛小鼠表皮和真皮中的 T 细胞均显著减少(图 4B,C)。尽管巨噬细胞和中性粒细胞数量未观察到统计学显著差异,但 H₂给药呈现出减少趋势,提示慢性炎症整体减轻(图 4D,E)。
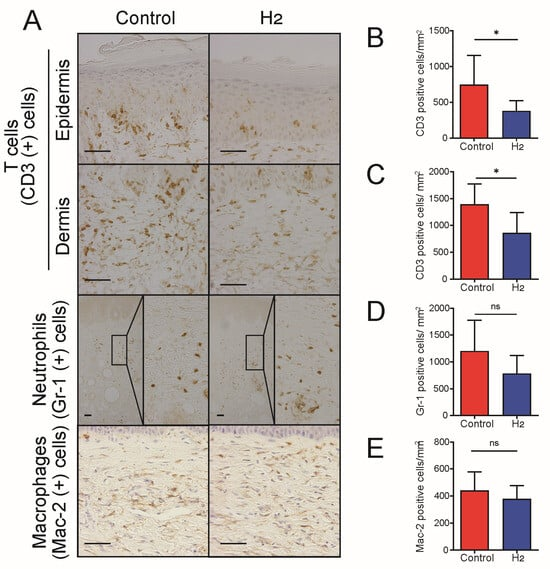
图 4. H₂治疗减少慢性 UVB 暴露后皮肤中的 T 细胞浸润。(A)CD3⁺ T 细胞、Gr‑1⁺ 中性粒细胞和 Mac‑2⁺ 巨噬细胞在表皮和真皮中的免疫组化染色。(B,C)CD3⁺ 细胞数量的定量分析显示 H₂治疗后显著减少。(D,E)Gr‑1⁺ 中性粒细胞和 Mac‑2⁺ 巨噬细胞的定量分析显示中性粒细胞浸润无显著变化,巨噬细胞积累略有减少。数据以 mean ± SD 表示(n=7)。*p<0.05;ns 表示无显著差异。比例尺 = 50 μm。
随后,我们使用实时 PCR 定量 UVB 照射 10 周后无毛小鼠背部皮肤中炎症介质的表达。比较 H₂治疗组和对照组中四种关键炎症介质——TNF‑α、IL‑1β、IL‑6 和 Ptgs2 的表达水平。然而,这些标志物均未观察到显著降低。这一出乎意料的结果可能归因于反复刺激后表观遗传修饰介导的细胞因子抵抗[22]或冗余通路激活[23],即使在 H₂治疗下仍维持慢性炎症(图 S5A–D)。
为更好地理解 H₂的抗炎作用,我们检查了单次 UVB 照射后 8 小时收集的皮肤样本,并证实 H₂在急性期抑制了这些炎症介质的表达。IL‑6 表达显著降低,IL‑1β 呈现降低趋势,与先前报道的 H₂抗炎作用一致(图 S5E–H)。
2.5 H₂减轻长期 UVB 照射皮肤中的 STAT3 激活和 IL‑6 产生
STAT3 激活与炎症相关的 UVB 诱导皮肤癌发展密切相关[24]。为研究 H₂给药后皮肤中 STAT3 激活的变化,我们通过免疫组化染色检查了 UVB 照射 10 周后收集的背部皮肤样本中 UVB 诱导的磷酸化 STAT3 核转位(图 5A,B)。核阳性细胞的定量比较显示,H₂治疗的无毛小鼠 STAT3 激活显著低于对照组。此外,Western blot 分析独立证实了 H₂治疗小鼠皮肤中 STAT3 激活的降低(图 5C,D)。
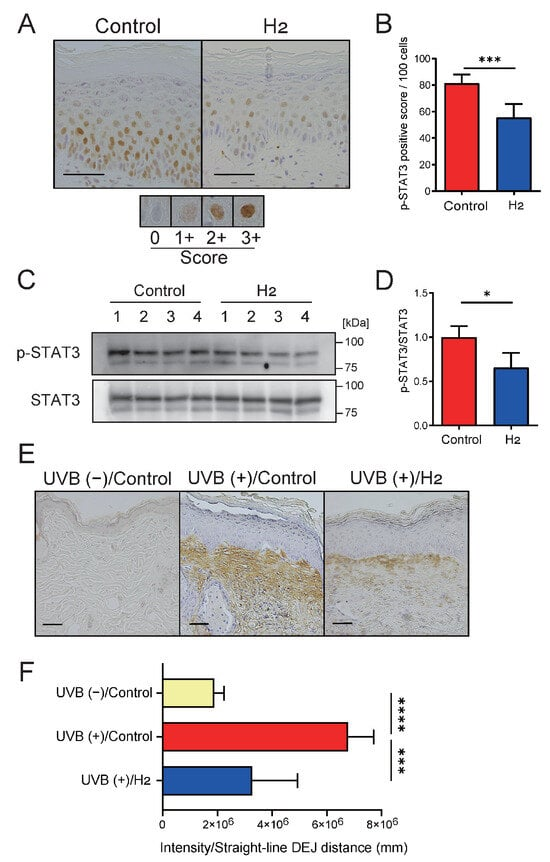
图 5. H₂治疗抑制信号转导和转录激活因子 3(STAT3)的激活,并限制白细胞介素‑6(IL‑6)的空间分布。
(A)磷酸化 STAT3(p‑STAT3)核定位的免疫组化染色。图中显示了免疫染色的评分标准。(B)定量分析显示 H₂治疗后 p‑STAT3 阳性细胞核减少(n = 7)。(C)p‑STAT3 和总 STAT3 水平的免疫印迹分析。(D)密度定量分析表明 H₂治疗后 p‑STAT3/STAT3 比值降低。(E)UVB(‑)/对照组、UVB(+)/对照组和 UVB(+)/H₂治疗组皮肤的 IL‑6 免疫组化染色。UVB 暴露诱导广泛的表皮下 IL‑6 阳性区域,而 H₂治疗显著减少该区域。(F)真皮‑表皮交界(DEJ)下方 IL‑6 染色强度的定量分析显示,H₂治疗显著减弱了 UVB 诱导的 IL‑6 增加(n = 4–7)。比例尺 = 50 μm。数据以 mean ± SD 表示。* p < 0.05;* p < 0.001; p < 0.0001。
接下来,我们检测了皮肤中 STAT3 激活的主要上游调节因子 IL‑6 的表达。图 5E 显示,对照组样本的皮下组织中广泛存在 IL‑6 免疫反应性,与先前报道一致[25]。H₂治疗显著减少了皮下组织局部区域的 IL‑6 免疫反应性。对真皮‑表皮交界下方 IL‑6 阳性染色区域的定量比较显示,H₂治疗组显著减少(图 5F)。尽管这些变化在 mRNA 水平未得到证实,但 H₂给药后慢性 UVB 照射皮肤中 IL‑6 蛋白产生减少,这可能削弱了 STAT3 的激活。或者,H₂可能直接削弱由 IL‑6 以外的刺激诱导的 STAT3 激活,两种可能性均成立。
2.6. H₂减轻长期 UVB 照射皮肤中的 ERK 和 JNK 信号通路激活
鉴于 H₂给药可减少炎症并削弱特定的细胞内信号通路[11,26,27],我们在 STAT3 之外进一步分析了其他关键信号分子的激活状态。具体而言,我们通过 Western blot 检测了 UVB 照射 10 周后皮肤样本中 ERK、p38 丝裂原活化蛋白激酶(p38)、JNK 和 AKT 的磷酸化水平(图 6A–F)。条带强度的定量分析证实,H₂治疗组的 ERK 和 JNK 激活显著降低,而 p38 和 AKT 的磷酸化水平保持不变。
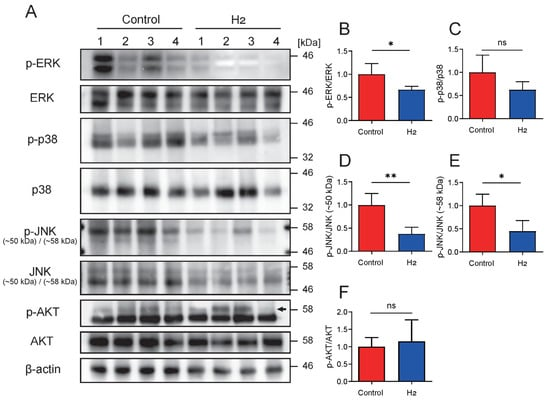
图 6. H₂选择性抑制 ERK 和 JNK 的磷酸化,而不影响 p38 或 AKT。
(A)p‑ERK/总 ERK、p‑p38/总 p38、p‑JNK/总 JNK(约 50 kDa 和 58 kDa 两条带)以及 p‑AKT/总 AKT 的代表性免疫印迹,β‑actin 作为内参。显示了分子量标记(kDa);箭头指示用于定量 p‑AKT 的 AKT1 条带。(B–F)ERK(B)、p38(C)、JNK(约 50 kDa)(D)、JNK(约 58 kDa)(E)和 AKT(F)的磷酸化/总蛋白比值的密度定量分析。H₂治疗显著降低 p‑ERK 和 p‑JNK 水平,而 p‑p38 和 p‑AKT 保持不变。数据以 mean ± SD 表示。* p < 0.05; p < 0.01;ns 表示无显著差异。
2.7. H₂减轻慢性 UVB 照射皮肤的增厚并抑制细胞增殖
我们检测了 UVB 照射 10 周后皮肤的表皮厚度和细胞增殖情况。与细胞内信号通路被抑制的结果一致,H₂治疗组的表皮厚度显著低于对照组(图 7A,B)。此外,通过 Ki‑67(图 7A,C)和 PCNA(图 7A,D)免疫染色评估的增殖细胞数量在 H₂治疗组中显著减少。
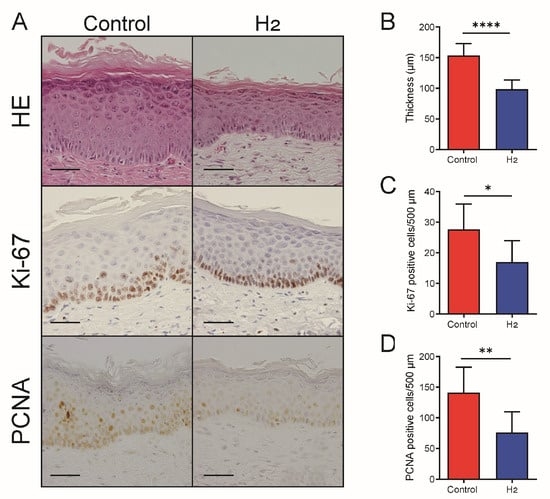
图 7. H₂减轻慢性 UVB 照射后的表皮增厚和细胞增殖。
(A)HE、Ki‑67 和 PCNA 免疫染色的代表性图像显示,H₂治疗样本的表皮厚度减少,增殖细胞数量降低。比例尺 = 30 μm。(B)表皮厚度的定量分析。(C)每 500 µm 中 Ki‑67 阳性细胞的定量分析。(D)每 500 µm 中 PCNA 阳性细胞的定量分析。与对照组相比,H₂治疗显著降低表皮厚度和增殖指数。数据以 mean ± SD 表示(n = 7)。* p < 0.05; p < 0.01; p < 0.0001。
2.8. H₂抑制急性和慢性 UVB 暴露后的 ROS 产生
在多种动物疾病模型中,H₂给药可持续降低 ROS 水平。因此,本研究中我们探讨了 H₂在 UVB 诱导皮肤癌模型中对 ROS 产生的影响。首先,我们在 H₂预处理 3 周后,检测了单次 UVB 照射对皮肤 ROS 水平的影响,并使用 GSH/GSSG 比值作为氧化应激的定量指标。在未给予 H₂的对照组中,UVB 照射后 GSH/GSSG 比值显著下降,这是由 ROS 产生引起的。相比之下,H₂给药组未观察到 GSH/GSSG 比值的显著下降,证实了 H₂对 UVB 诱导皮肤 ROS 产生的抑制作用(图 8A)。
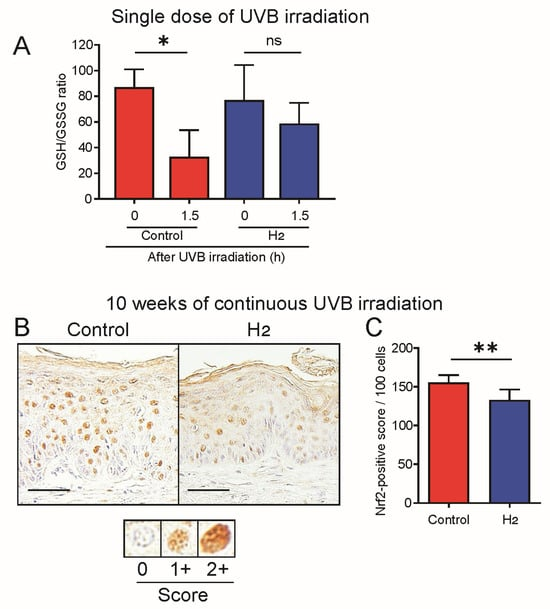
图 8. H₂降低急性和慢性 UVB 诱导皮肤损伤中的氧化应激标志物。
(A)急性 UVB 模型中的 GSH/GSSG 比值。单剂量 UVB 照射在 1.5 h 时降低了对照组小鼠的 GSH/GSSG 比值,而 H₂预处理维持了该比值(n = 5–6)。(B)慢性 UVB 模型(10 周)中核 Nrf2 的代表性免疫组化图像。(C)每 100 个细胞中核 Nrf2 阳性评分的定量分析。H₂治疗减少了核 Nrf2 的积累,表明 ROS 诱导的激活减少(n = 6–7)。比例尺 = 50 μm。数据以 mean ± SD 表示。* p < 0.05; p < 0.01;ns 表示无显著差异。
为评估 H₂对慢性 UVB 暴露皮肤中 ROS 产生的影响,我们首先比较了作为脂质过氧化标志物的丙二醛水平。然而,H₂治疗组和对照组之间未观察到显著差异,这很可能是由于 10 周内每周 UVB 照射产生的大量 ROS 导致脂质氧化产物长期累积所致。
由于 ROS 激活的 Nrf2 核转位是一个可逆过程,大约在一天内下降[28,29],我们使用 Nrf2 的核转位作为动态指标来评估短期 ROS 周转情况。如图 8B 所示,与对照组相比,H₂治疗组的 Nrf2 核转位明显减少。核转位信号的定量分析证实,H₂治疗组的核 Nrf2 积累显著降低(图 8C)。
3. 讨论
我们的研究表明,通过饮用 HRW 和吸入氢气持续给予 H₂约六个月,可减少无毛小鼠中 UVB 诱导的皮肤肿瘤累积数量。该治疗显著延迟了早期病变的出现,减少了肿瘤数量,并延长了生存时间。尽管在初始队列中 SCC 的发生未被显著延迟,但在独立重复实验中观察到了显著延迟。尽管研究人员先前已在人类和小鼠模型中将 H₂给药与 UV 照射结合,但这些研究主要关注短期 UV 暴露后的急性反应[18],测量 ROS 减少或炎症细胞因子水平等即时结果[30,31,32],而未研究致癌过程。长期研究仍局限于两篇报道:Kiyoi 等人发现六周的 H₂治疗可抑制 UVA 诱导的小鼠表皮增生和黑色素生成[33],而 Kato 等人报道人类三个月氢浴后皱纹减少[34]。
除角质形成细胞为主的模型外,H₂还对人黑素细胞具有细胞保护作用。例如,Fang 等人证明 H₂通过激活 Nrf2 信号通路减少氧化应激并保护人黑素细胞[35]。此外,在一项初步人体研究中,局部应用 HRW 改善了包括色素相关指标在内的客观皮肤参数[36]。这些观察结果表明,H₂可以在与 UV 驱动皮肤生物学相关的氧化应激条件下调节氧化还原和色素相关通路。尽管我们目前的工作关注 UVB 诱导的角质形成细胞癌变,但氧化应激和 Nrf2 相关信号在角质形成细胞和黑素细胞中的共同参与值得未来使用黑色素瘤相关模型进行研究,以直接测试 H₂是否影响黑素细胞转化或黑色素瘤的发生/进展。
多项动物研究表明,H₂给药可在多种疾病模型中改变致癌过程,而不仅限于 UV 诱导的皮肤癌。在非酒精性脂肪性肝炎(NASH)进展为肝细胞癌的小鼠模型中,饮用 HRW 八周与对照组相比显著降低了肝肿瘤发生率和结节大小[37]。同样,在铁‑次氮基三乙酸(Fe‑NTA)诱导的大鼠化学致癌模型中,饮用 HRW 可抑制肾脏早期肿瘤促进事件[38]。此外,在辐射诱导的致癌小鼠模型中,照射前给予富氢盐水与对照组相比显著降低了胸腺淋巴瘤的发生率[39]。总体而言,这些研究表明 H₂的抗癌作用主要归因于其抗氧化活性和氧化应激减少。
在我们的研究中,对 UVB 照射的无毛小鼠给予 H₂可延迟早期 SCC 病变的出现;然而,在照射后 30 周,组织学类型分布、分化等级和浸润深度无显著差异。在我们的给药条件下,UVB 诱导的 CPD 形成在两组之间没有差异。相比之下,在人类皮肤中,UV 照射前局部应用 H(H₂O)m(一种“由水分子包围的原子氢”制剂)可通过 UV 诱导的红斑和胸腺嘧啶二聚体形成评估抑制 DNA 损伤[18]。与通过吸入或自由饮用给予 H₂相比,H(H₂O)m 可能为基因组提供更强的 DNA 损伤保护。此外,间歇性 H₂吸入减少了 UVA 照射诱导的 8‑OHdG(氧化 DNA 损伤标志物)形成[33]。8‑OHdG 是 ROS 引起的 DNA 氧化损伤的生物标志物[40],尽管我们的研究未测量该指标。考虑到 H₂给药的氧化应激降低作用,我们的无毛小鼠模型有望得到类似结果。总体而言,通过吸入气体或 HRW 给予的 H₂可能不会直接干扰 UV 光产物形成、突变获得或克隆选择。相反,它可能通过调节肿瘤微环境(包括炎症反应和细胞内信号)并减少氧化应激来影响肿瘤形成。
H₂不经历常规的酶代谢;相反,其在体内的命运以快速全身扩散、消除和部分组织消耗为特征。摄入 HRW 后,呼出 H₂浓度在几分钟内升高,并在约 45–60 分钟内恢复到基线[41]。一项人体质量平衡研究表明,约 59% 的摄入 H₂通过呼气排出,约 0.1% 通过体表排出,而剩余约 40% 未被回收,推测保留在组织中或在氧化条件下被消耗[42]。
与体内部分消耗一致,低水平吸入(160 ppm)期间的全身 H₂消耗量约为 0.7 μmol/min/m² 体表面积[43]。药代动力学特征因给药途径而异。在啮齿动物中,口服 HRW 可使肝脏和静脉血中的 H₂浓度快速但短暂升高,而动脉水平仍然显著较低;相比之下,吸入可使静脉和动脉血中的 H₂浓度类似升高[44]。支持这种途径依赖性分布的是,一项猪研究表明,富氢溶液的空肠给药增加了门静脉 H₂浓度,而颈动脉水平仍未检测到,表明在全身分布之前存在快速的首过肝清除和/或肺消除[45]。H₂的组织分布还因一个有趣的观察结果而更加复杂:据报道,口服给药后肝糖原可保留 H₂,表明肝脏可能作为短暂的储存库[46]。
这些药代动力学特征表明,HRW 提供短暂的脉冲暴露,而吸入导致更持续的全身可用性。因此,重复给药是长期调节炎症相关信号的合理方法[42,44]。先前的体内研究进一步表明,疗效取决于给药方案;例如,连续 2% 氢气吸入仅产生轻微效果,而间歇性 2% 氢气暴露和 HRW 提供更明确的保护,这与受益于反复短暂 H₂升高的信号调节机制一致[41]。基于这些观察,我们采用了连续 2% 氢气吸入以维持基线可用性,加上自由饮用 HRW 以叠加间歇性脉冲的联合方案,从而补偿快速清除并确保在整个慢性 UVB 暴露期间持续的 H₂可用性[42,44]。
H₂给药可抑制动物疾病模型中的 ROS 产生。在本研究中,我们评估了皮肤中的 ROS 抑制情况,发现在 UVB 照射 10 周的无毛小鼠中,H₂给药显著减少了 Nrf2 的核定位。单次 UVB 暴露在无毛小鼠皮肤中诱导氧化应激,而 H₂给药有效减少了这种氧化应激。除了直接清除羟自由基和过氧亚硝酸盐[10]外,H₂还通过兴奋效应机制激活 Nrf2 发挥抗氧化作用[47]。Nrf2 敲除小鼠的研究表明,Nrf2 激活介导了 H₂的抗炎作用[48],确立了 Nrf2 作为 H₂抗氧化和抗炎作用的关键分子[49]。最近,Fe‑卟啉被确定为 H₂的分子靶点[50],羟自由基对 Fe(II)‑血红素的氧化产生 Fe(III)–OH 加合物,该加合物可与 H₂反应形成 Fe(III)–H 氢化物物质。这些加合物可能氧化 Keap1 传感器结构域中的半胱氨酸残基,从而释放并激活 Nrf2[51]。
最近的证据进一步表明,除了对羟自由基和过氧亚硝酸盐等高反应性氧化剂的拟议反应性外,H₂还可以与氧化还原活性金属蛋白相互作用。特别是,线粒体电子传递链复合物 III 的催化亚基 Rieske 铁‑硫蛋白(RISP)已被确定为 H₂的主要分子靶点[52]。H₂激活线粒体 Lon 肽酶 1(LONP1)并促进 RISP 降解,从而诱导线粒体未折叠蛋白反应(UPRmt)。结构分析表明,Rieske 中心的 [2Fe–2S] 簇由两个半胱氨酸和两个组氨酸残基配位;值得注意的是,组氨酸配体暴露于溶剂,可能允许小分子访问该氧化还原辅因子。这些发现,连同 Fe‑卟啉可参与 H₂响应性氧化还原化学的证据,支持了一种模型,其中 H₂通过调节金属中心的氧化还原化学和氧化剂敏感信号而非传统受体结合来影响致癌过程。在 UVB 诱导的皮肤癌发生中,这种氧化还原调节可能减少氧化应激并减弱下游促炎和促增殖通路,包括 IL‑6/STAT3 和 ERK/JNK,从而优先影响肿瘤促进而非 UV 光产物驱动的起始阶段。
尽管当细胞在富含 H₂的环境中遇到 ROS 时,H₂可以诱导快速的 Nrf2 激活,但长期 ROS 产生加上持续 H₂暴露可能建立不同的平衡。与此一致,在富含 H₂的培养基中培养的人脂肪组织显示 Nrf2 表达降低[53]。因此,在 H₂治疗的皮肤中观察到的 Nrf2 活性降低可能反映了 UV 诱导的 ROS 产生与 H₂/Nrf2 介导的清除之间的平衡,从而实现高效的 ROS 去除并减少对 Nrf2 激活的需求。
我们的观察结果表明,UVB 同时在皮肤中产生 ROS 并造成直接 DNA 光损伤。这些 ROS 通过诱导角质形成细胞和免疫细胞产生 IL‑1β、IL‑6 和 TNF‑α 建立慢性炎症微环境。这种炎症反应驱动 SCC 的发生和进展[5]。我们实验中 UVB 诱导 SCC 的延迟出现可能是由于 H₂介导的 ROS 减少从而减轻炎症,或者是由于 H₂的直接抗炎特性阻止了初始病变的形成。与此框架一致,抗氧化干预(例如富含多酚的黑莓提取物)可降低 COX‑2、iNOS、PGE₂,并抑制 MAPK(ERK、JNK、p38)和 NF‑κB/p65,从而减轻 UV 诱导的皮肤损伤[54]。同样,在 PM2.5 诱导的肺炎模型中,补充细胞内谷胱甘肽的 NAC 给药发挥强效抗氧化作用,减少 ROS 依赖性中性粒细胞和单核细胞气道浸润,并减轻炎症[55]。过量的 ROS 激活 NF‑κB 转录因子,诱导 IL‑1β、TNF‑α 和黏附分子的表达[56]。UVB 辐射强烈激活角质形成细胞中的 ERK/JNK,通过增强细胞增殖、逃避凋亡和激活 AP‑1 直接促进肿瘤发生[57]。此外,ROS 刺激 MAPK 通路[58]和 NLRP3 炎症小体[59],从而放大炎症介质的产生。因此,我们假设 H₂介导的 ROS 减少可减轻 UV 照射上皮中的炎症和细胞内信号,从而减少上皮增生和增殖。因此,H₂可能延迟肿瘤发展。
H₂给药显著减少了慢性 UVB 暴露后 T 细胞向表皮和真皮的浸润,限制了 IL‑6 蛋白在皮下组织的分布,并降低了 STAT3 的核转位。由于异常的 STAT3 激活广泛参与包括皮肤 SCC 在内的上皮肿瘤发生,并且其组成性磷酸化会上调促存活和促增殖基因,因此这些变化在机制上与肿瘤发展延迟一致[60]。H₂似乎通过两种机制抑制 UV 诱导的慢性炎症:一是减少氧化应激,二是抑制炎症相关信号通路,特别是 IL‑6/STAT3 和 ERK/JNK 级联反应。总体而言,这些作用削弱了由 UV 诱导的慢性炎症触发的 SCC 早期发展阶段,从而延迟乳头状瘤的形成并减少肿瘤数量。尽管在慢性阶段炎症细胞因子的 mRNA 表达未显示显著改变,但在急性 UVB 暴露后 8 小时观察到 IL‑6 的明显抑制。这表明对急性反应的累积抑制最终表现为慢性炎症期间蛋白水平的变化。
巨噬细胞是另一个能够塑造 UV 暴露皮肤微环境的免疫细胞群。在本研究中,Mac‑2⁺ 巨噬细胞的浸润水平较低,并且在对照组和 H₂组之间没有显著差异,表明在我们的实验条件下,H₂并未显著改变巨噬细胞的总数量。然而,巨噬细胞的极化和功能可以独立于细胞总数发生变化;事实上,最近在皮肤伤口模型中的研究表明,吸入氢气可促进早期 M2 样极化并减轻炎症[61,62]。因此,H₂可能影响巨噬细胞的表型和细胞因子输出,而不是其募集。未来的研究应使用流式细胞术或多重免疫染色来表征巨噬细胞亚群(例如 iNOS/CD86 与 Arg1/CD206),以确定在 UVB 驱动的肿瘤促进过程中,巨噬细胞来源的细胞因子是否参与 IL‑6/STAT3 信号传导。除了光致癌作用外,这些免疫调节特性也可能与特应性皮炎、痤疮或银屑病等炎症性皮肤病相关,尽管还需要进一步的对照研究来证实。
在两项独立研究中,H₂给药将 UVB 诱导的早期 SCC 病变出现时间平均延迟了 3 周。比较小鼠和人类衰老的研究表明,17 周龄小鼠的肿瘤形成速度比人类快 45 倍[63],这意味着小鼠的 3 周延迟相当于人类约 2 年 7 个月。尽管跨物种的衰老和肿瘤发生换算只是近似值,但这些发现表明,H₂给药可能有助于在人群水平上降低与年龄相关疾病的发病率。
UV 诱导皮肤肿瘤的早期病变存在物种特异性差异:小鼠通常会形成乳头状瘤[64],而人类则表现为癌前上皮内病变,例如光化性角化病(AK)[65]和鲍温病(也称为原位鳞状细胞癌)[66]。因此,在将小鼠数据外推至人类疾病时需要谨慎解释。这些差异可能由多种相互作用的因素造成:皮肤结构差异[67]、起源细胞不同[68,69]、UV 暴露方案差异[70]、遗传背景不同[71]以及早期驱动突变的谱和功能差异。人类中常见早期 TP53 和 NOTCH 功能丧失,与分化缺陷相关[72],而小鼠 UVB 模型则主要表现为表皮增生和增殖增加[73]。因此,能够更好地模拟人类 UV 诱导 SCC 的小鼠模型——在 UV 光谱、遗传背景和靶细胞方面与人类更接近——将能更准确地评估 H₂对人类皮肤 SCC 发展的影响。
本研究存在一些局限性。首先,H₂以联合方案给药(连续 2% 吸入 + 自由饮用 HRW),未设置单独吸入、单独 HRW 或间歇性吸入的对照组。此外,我们没有直接量化血液、皮肤或组织中的 H₂浓度。因此,给药剂量是通过舱内 H₂浓度(2 vol%)和饮用水中的溶解 H₂浓度(≥0.8 mM)来操作性定义的。先前的药代动力学研究表明,HRW 会导致组织 H₂快速但短暂升高,而吸入则能产生更稳定的血液水平[44,74]。由于不同给药途径可能导致组织 H₂水平不同,未来的研究必须直接测量靶组织中的 H₂浓度,以阐明途径和模式特异性的暴露特征,并将局部 H₂可用性与 IL‑6/STAT3 和 ERK/JNK 信号的调节联系起来。其次,我们的评估主要集中在肿瘤早期发展,而对已建立的 SCC 的增殖、侵袭、转移以及肿瘤免疫微环境的影响尚未探索。先前的细胞系研究表明,当线粒体功能较高时,补充 H₂可能促进细胞增殖[75]。最近的研究已将 RISP 确定为 H₂的主要靶点,并提出 RISP 缺失及其后的 UPRmt 诱导是 H₂多效性和环境依赖性作用的基础[52]。这为看似矛盾的观察结果提供了潜在的机制解释;因此,未来的研究应在连续 H₂给药条件下,评估 UVB 暴露皮肤中的线粒体蛋白稳态、UPRmt 标志物以及肿瘤生长动态。第三,SCC 发生的延迟未达到统计学显著性,并且在重复性实验中生存延长也处于临界水平。这可能取决于统计效力或事件定义。第四,本模型仅限于雄性 HR‑1 无毛小鼠的单纯 UVB 暴露,这限制了其对人类 UVA/UVB 混合暴露以及通过 AK/鲍温病发生癌变的外推有效性。第五,观察到的 IL‑6/STAT3 和 ERK/JNK 抑制仅为相关性发现,通过抑制剂或基因干预验证因果关系仍未完成。第六,尽管 DNA 光产物(CPD)未发生变化,但我们未对氧化 DNA 损伤或突变负荷进行全面分析。基于这些考虑,未来的研究应解决途径特异性给药并进行暴露测量,评估对已建立肿瘤的影响,在包含 UVA 的条件下在雌性和其他品系中重复实验,并使用干预方法进行机制验证。
4. 材料与方法
4.1. 实验动物
4 周龄雄性无毛小鼠(Hos:HR‑1)购自日本 SLC(滨松,日本)。小鼠饲养在塑料笼中,环境条件控制为 22 °C ± 2 °C、相对湿度 50% ± 10%,并保持 12 小时光照‑黑暗周期(08:00 开灯),自由采食标准啮齿动物饲料(CRF‑1;东方酵母株式会社,东京,日本)。如图 1 所示,小鼠从 4 周龄至 27 周龄接受 H₂治疗,并从 7 周龄至 27 周龄接受背部皮肤 UVB 照射。每周肉眼观察并记录皮肤肿瘤的形状、大小和数量。所有实验程序均获得中部大学动物实验委员会批准(批准号:2410011,批准日期:2024 年 3 月 12 日),并符合美国国立卫生研究院(NIH)《实验动物护理和使用指南》。
4.2. H₂给药和 UVB 照射
小鼠被分为 H₂组或对照组。所有组别均在相同环境条件下饲养,实验操作在固定时间进行,以尽量减少昼夜节律影响。H₂组同时接受 2% 氢气吸入和自由饮用 HRW,而对照组接受空气和去氢水。由于口服 H₂在体内清除迅速,其生物学效应取决于剂量方案和给药途径[44],因此我们采用了连续 2% 氢气吸入 + 自由饮用 HRW 的联合方案。该设计旨在通过吸入维持稳定的全身 H₂水平,同时通过饮水提供间歇性脉冲暴露,这种模式在其他体内模型中被报道比单独连续吸入产生更显著的生物学效应[41,42,43]。氢气按照先前建立的方案[44]以含 H₂空气的形式在丙烯酸舱内给药。简要来说,将标准塑料笼中的小鼠置于密封的 60 L 丙烯酸舱内,持续通入 2% 氢气/98% 空气混合气体,流速为 10 L/min。混合气体通过将 100% 氢气(太阳日酸,东京,日本)与涡旋式压缩机提供的压缩空气(SLP‑15EB;阿耐思特岩田,横滨,日本)混合产生,并通过多流量计(Model‑1203;Kofloc,京都,日本)调节。对照组小鼠在相同条件下饲养,但通入不含氢气的空气,流速相同。定期收集舱内空气样本,并使用光学气体监测仪 Model FI‑21(理研计器,东京,日本)监测 H₂浓度,方法如前所述[44]。为确保稳定的 H₂暴露,HRW 每天(周末除外)使用 Aquela Hydrogen Water 7.0(由 MiZ 株式会社捐赠,镰仓,日本)制备。制备后使用氢电极(ABLE,东京,日本)确认溶解 H₂浓度(≥0.8 mM),并每 24 小时更换水瓶以尽量减少溶解 H₂的损失。去氢水通过将 HRW 在 4 °C 下敞口放置 48 小时获得。
小鼠从 7 周龄至 27 周龄,使用两支 G8T5E UVB 灯(三共电机,平冢,日本)对背部皮肤进行 UVB 照射,剂量为 270 mJ/cm²,每周 3 次,共 20 周。UVB 照射剂量会影响皮肤损伤的严重程度和肿瘤发展速度[76]。基于无毛小鼠中已建立的光致癌方案,我们选择每次照射剂量为 270 mJ/cm²。Epstein 等人的研究表明,每周 3 次、每次 270 mJ/cm² 的 UVB 照射可有效诱导肿瘤[77]。先前的研究在无毛小鼠皮肤致癌模型中也采用了每周 3 次、每次 240–250 mJ/cm² 的剂量[78,79]。该剂量相当于 1–2 个最小红斑量(MED)[78,80],并且我们也观察到了早期红斑形成(图 S1)。该方案旨在在实验期间可靠地诱导 UVB 介导的病变和肿瘤,同时符合人道终点标准。在照射后 10 周的观察期结束时,小鼠在 37 周龄时被安乐死以进行组织学检查。每周至少监测一次小鼠的肿瘤发展、体重和整体健康状况。肿瘤最大直径不允许超过 15 mm;在实际操作中,当肿瘤最大直径达到约 10–15 mm 时,根据肿瘤位置和临床状况对小鼠实施安乐死。人道终点根据机构指南确定。当出现以下任一情况时实施安乐死:体重较基线下降超过 20%;肿瘤出现溃疡、坏死、感染或持续出血;或肿瘤生长导致活动、进食、饮水或呼吸受损。
4.3. CPD 测量
使用 PureLink Genomic DNA Mini Kit(Invitrogen,沃尔瑟姆,马萨诸塞州,美国)从 5 mm 见方的小鼠背部皮肤中提取基因组 DNA。使用 High-Sensitivity CPD ELISA Kit Ver. 2(Cosmo Bio,东京,日本)按照制造商的方案定量 CPD 水平。
4.4. 组织学和免疫组化分析
皮肤或肿瘤组织在 10% 中性缓冲福尔马林中固定 48 小时,随后脱水并石蜡包埋。制备 4 µm 厚切片用于 HE 染色和免疫组化。组织病理学诊断由两位病理学家根据 HE 染色切片共同确定。免疫组化按照我们已建立的方案[81]进行,使用以下一抗:抗 CD3(Dako,卡平特里亚,加利福尼亚州,美国)、抗 Gr‑1(eBioscience,圣地亚哥,加利福尼亚州,美国)、抗 Mac‑2(Cedarlane,安大略省,加拿大)、抗 IL‑6(Novus Biologicals,森特尼尔,科罗拉多州,美国)以及抗 STAT3、Ki‑67、PCNA 和 Nrf2 的抗体(Cell Signaling Technology,丹弗斯,马萨诸塞州,美国)。
4.5. RNA 提取和逆转录定量 PCR(RT‑qPCR)
使用 RNeasy Mini Kit(Qiagen,希尔德,德国)提取总 RNA,并使用 Transcriptor First-Strand cDNA Synthesis Kit(Roche,巴塞尔,瑞士)将 2 µg RNA 逆转录为第一链 cDNA。qPCR 步骤使用 LightCycler 仪器和 FastStart DNA MasterPLUS SYBR Green I(Roche,巴塞尔,瑞士)进行。用于扩增的引物如下:IL‑6(正向:5′‑TCCCAACAGACCTGTCTATACC‑3′;反向:5′‑CAGAGGAAATTTTCAATAGGCA‑3′);IL‑1β(正向:5′‑TCCTCTCCAGCCAAGCTTCC‑3′;反向:5′‑TTGATGTGCTGCTGCGAGATT‑3′);TNF‑α(正向:5′‑AACTTCGGGGTGATCGGTCC‑3′;反向:5′‑GCAAATCGGCTGACGGTGTG‑3′);以及 Ptgs2(正向:5′‑TTCCAATCCATGTCAAAACCGT‑3′;反向:5′‑GGGGTGGGCTTCAGCAGTAA‑3′)。所有步骤均如前所述进行[44]。
4.6. 免疫印迹分析
蛋白质裂解物的 Western blot 分析如前所述进行[44]。膜与识别 p38 MAPK、SAPK/JNK、ERK1/2 和 AKT 的磷酸化形式和总形式的一抗孵育(Cell Signaling Technology,贝弗利,马萨诸塞州,美国)。β‑Actin(Sigma,圣路易斯,密苏里州,美国)作为内参。分子量标记使用 Blue Prestained Protein Standard, Broad Range(11–190 kDa;NEB P7706S;New England Biolabs,伊普斯威奇,马萨诸塞州,美国)。免疫反应条带使用 ECL Prime Western Blotting Detection Reagent(GE Healthcare,芝加哥,伊利诺伊州,美国)可视化,用 Fusion Solo S 系统(Vilber Bio Imaging,科勒吉恩,法国)成像,并使用 ImageJ 软件(v. 1.53k)进行定量。
4.7. 皮肤中 GSH 和 GSSG 的定量
使用 GSSG/GSH Quantification Kit(Dojindo,熊本,日本)测定皮肤样本中的还原型和氧化型谷胱甘肽浓度。
4.8. 统计分析
所有统计分析均使用 GraphPad Prism 版本 10.6.1(GraphPad Software,波士顿,马萨诸塞州,美国)进行。根据情况,使用 Student’s t 检验、单因素 ANOVA 结合 Tukey 事后检验或双因素 ANOVA 结合 Šidák 事后检验分析组间差异。分类变量使用卡方检验比较。使用 Kaplan–Meier 法估计肿瘤发生率和生存率,并通过 log‑rank 检验比较差异。使用 Cox 比例风险回归估计风险比和 95% 置信区间。数据以 mean ± SD 表示。统计学显著性定义为双侧 p 值 < 0.05。
5. 结论
在 HR‑1 无毛小鼠中,持续给予 H₂可延迟 UVB 诱导皮肤癌发生的早期炎症阶段,并且这一结果在独立队列中得到验证。尽管 CPD 水平未发生变化,但该干预减轻了皮肤中的 IL‑6/STAT3 和 ERK/JNK 信号传导,维持了氧化还原平衡(急性 UVB 暴露后 GSH/GSSG 升高,慢性暴露期间核 Nrf2 降低),并抑制了表皮增殖和增厚。这些结果表明,在慢性 UVB 照射下,H₂主要调节肿瘤促进阶段而非起始阶段,这解释了观察到的乳头状瘤发生延迟。这些发现为进一步研究 H₂作为 UV 照射皮肤的化学预防剂提供了依据。
https://blog.sciencenet.cn/blog-41174-1518124.html
上一篇:开放科学运动真的如支持者所言那般有益吗?
下一篇:AI 模型竟然会患心理疾病!