博文
氢气2%吸入对体外心肺复苏临床试验方案【哈佛】
||
氢气2%吸入对体外心肺复苏临床试验方案【哈佛】
摘要
背景
体外心肺复苏(ECPR)可提高难治性心脏骤停患者的存活率,但常伴随严重的缺血再灌注损伤(IRI)。氢气(H₂)在减轻缺血再灌注损伤方面已展现出良好的临床前疗效。“Hydrogen-FAST试验”旨在研究先天性心脏病(CHD)患者在ECPR期间吸入氢气作为辅助治疗的可行性与安全性。
方法
本项1期多中心随机对照试验纳入了因潜在心脏疾病相关的难治性心脏骤停而接受ECPR治疗的所有年龄段患者。由于ECPR事件具有紧急性,该试验采用“知情同意豁免”纳入流程。53名受试者将被随机分为两组:一组接受标准治疗,另一组接受标准治疗联合氢气吸入(通过机械通气和体外膜肺氧合(ECMO)回路持续输送2%浓度的氢气,时长72小时)。主要终点包括:①可行性——以心脏骤停后最初72小时内氢气成功输送的时间百分比衡量,可行性定义为平均百分比≥90%;②安全性——通过治疗相关严重不良事件(SAEs)的发生率评估。次要终点则考察临床结局,包括出院存活率、ICU住院时长、6个月时的功能状态、神经功能及缺血损伤标志物。
讨论
Hydrogen-FAST试验将为ECPR期间氢气给药的可行性和安全性提供关键数据,为更大规模的疗效研究以及氢气在危重症监护场景中潜在的广泛临床应用提供参考。
Trial registration
ClinicalTrials.gov NCT05574296. Registered on October 6, 2022. https://clinicaltrials.gov/study/NCT05574296.
Administrative information
Primary registry and trial identifying number {4} | ClinicalTrials.gov, NCT05574296. Registered on 6 October 2022 |
Secondary identifying numbers | Not applicable |
Source(s) of monetary or material support | National Heart, Lung, and Blood Institute (NHLBI), Grant numbers 1R33HL164373-01 and 1R61HL164373-01, and by the Mooney Family Foundation |
Primary sponsor and contact information {3b} | National Heart, Lung, and Blood Institute jewel.joshua@nih.gov 301–827−1809 |
Role of sponsor and funder {3c} | The study sponsors had no role in or ultimate authority over the study design; collection, management, analysis, and interpretation of data; writing of the report; and the decision to submit the report for publication |
Contact for public queries | https://research.childrenshospital.org/research-units/hydrogenfast-study-research hydrogenfast@childrens.harvard.edu |
Contact for scientific queries | John N. Kheir, MD (john.kheir@cardio.chboston.org) and Lynn A. Sleeper, ScD (lynn.sleeper@cardio.chboston.org) |
Public title | Hydrogen’s Feasibility and Safety as a Therapy in Extracorporeal Cardiopulmonary Resuscitation: Design of a Randomized Controlled Trial (Hydrogen-FAST Trial) |
Scientific title | Hydrogen’s Feasibility and Safety as a Therapy in Extracorporeal Cardiopulmonary Resuscitation: Design of a Randomized Controlled Trial (Hydrogen-FAST Trial) |
Countries of recruitment | USA |
Health condition(s) or problem(s) studied | Cardiac arrest with resuscitation using extracorporeal cardiopulmonary resuscitation (ECPR) |
Intervention(s) | Inhaled 2% hydrogen gas |
Key inclusion and exclusion criteria | Inclusion criteria (all must be fulfilled) will be: (1) Patients admitted to the cardiac intensive care unit at a participating site with cardiac comorbidity, including congenital heart disease, myocarditis, cardiac arrhythmia, or rejection of a transplanted heart (2) Patient experiencing a cardiac arrest >5 min and receiving ongoing CPR in the ICU, cardiac catheterization lab, or cardiac operating room (3) The decision made by the clinical team to resuscitate using ECPR Exclusion criteria will be (any one disqualifies patient): (1) Enrollment in the opt-out program (2) Patients known to be pregnant (3) Patients who are prisoners (4) Prior ECPR episode during admission (5) Enrollment does not occur within 6 h of the decision to resuscitate using ECPR (6) Patients enrolled in the Trial of Indication-Based Transfusion of Red Blood Cells in ECMO (TITRE) within the last 18 months |
Study type | Open-label randomized controlled trial |
Date of first enrollment | 29 March 2024 |
Sample size | 56 |
Primary outcome(s) | Primary feasibility endpoint. We will compute the percentage of the first 72 consecutive post-arrest hours (starting at the time of first CPR initiation) in which H2 gas was administered via all of the applicable pathways (e.g., mechanical ventilator and ECMO membrane) Primary safety endpoint. The primary safety endpoint will be the incidence rate of SAEs of interest per day during the first 30 days post-randomization that have been classified as treatment-related or possibly treatment-related as determined by the Adverse Event Adjudication Committee |
Key secondary outcome(s) | We will compute the percentage of the first 72 consecutive hours post-H2 initiation in which H2 gas was administered via all of the applicable pathways (e.g., mechanical ventilator and ECMO membrane) Survival to hospital discharge, ICU and hospital lengths of stay Functional status score (FSS) will be retrospectively computed at baseline (admission to the hospital and at 24 h before cardiac arrest), discharge (±3 days), and 6-month (±2 months) post-randomization Markers of ischemic injury, including lowest pH; highest lactic acid; lowest platelet count; and highest hepatic transaminases, blood urea nitrogen and creatinine, and international normalized ratio on post-randomization day 0, 1, 2, 3, 4, 5, 6, 7, 14, 21, and 28 Exploratory efficacy endpoints, including brain biomarkers (GFAP, Tau, and Neurofilament light) Findings on clinically obtained imaging (brain MRI, head CT, cranial ultrasound) |
Ethics review | Approved by the Institutional Review Board at Boston Children’s Hospital (IRB-P00043374) |
Individual trial participant data sharing statement | The sharing of deidentified individual clinical trial participant-level data is not applicable at this time |
引言
背景与理论依据{9a}
体外心肺复苏(ECPR)可改善难治性心脏骤停患者的预后[1,2],但该治疗常伴随严重的缺血再灌注损伤(IRI),进而导致患者发病与死亡。临床前研究表明,氢气可通过选择性清除羟自由基及调控炎症通路,减轻缺血再灌注损伤[3,4,5]。
对照方案选择说明{9b}
既往人体研究已证实氢气在心脏骤停治疗中的安全性[6,7]与有效性[8],但目前仍缺乏针对危重症人群的严谨评估。本试验将受试者分为两组:一组接受标准治疗联合2%氢气吸入,另一组仅接受标准治疗。
研究目的{10}
我们设计了一项多中心随机对照试验,旨在评估对接受ECPR治疗的婴幼儿及儿童患者给予氢气治疗的安全性与可行性,该试验全称为“氢气在ECPR中作为治疗手段的可行性与安全性研究(Hydrogen-FAST)”。本文将详细介绍该试验的设计方案。
方法:患者与公众参与及试验设计
患者与公众参与{11}
社区咨询
在各中心启动试验前,且在单一机构审查委员会(sIRB)批准后,研究团队会开展社区咨询与公开告知活动,确保相关社区人群了解并理解本试验。社区咨询包括与熟悉心脏重症监护室( cardiac ICU)环境的社区代表召开会议,就研究目的、研究流程、潜在风险及研究收益进行讨论。通过咨询收集的反馈可用于评估社区对“知情同意豁免(EFIC)”的态度、对研究风险与收益的理解程度、对参与研究的担忧,以及对研究流程的建议。研究团队还会单独与医护人员开展咨询,收集反馈意见,以完善公开告知流程与“退出试验(opt-out)”流程。咨询结果将进行汇总与定性分析,并连同公开告知计划一并提交至sIRB,待批准后方可启动试验。
公开告知
在获得sIRB批准后,研究团队会开展公开告知工作。各研究中心的公开告知计划可能存在差异,具体取决于当地社区咨询会议收集的反馈。例如,在波士顿儿童医院(Boston Children’s Hospital)与国家儿童医院(Children’s National Hospital),公开告知通过医院官网、社交媒体平台及新闻发布会开展,不仅提供试验相关详细信息与知情同意豁免流程说明,还会告知受试者退出试验的途径。此外,研究团队还会在重症监护室候诊区、诊疗区域及每个ICU病床旁张贴信息传单。
试验设计{12}
本研究为1期开放标签随机对照试验。随机分组(氢气治疗组与标准治疗组比例为2:1)将受试者分为两组:一组接受标准治疗联合2%氢气吸入,另一组仅接受标准治疗。当临床团队决定为患者行体外膜肺氧合(ECMO)插管时,即通过按年龄(<28天 vs ≥28天)与研究中心分层的密封信封进行随机分组。
试验场所{13}
本研究在美国多家儿童心脏疾病治疗中心开展。研究中心列表可在临床试验注册网站(ClinicalTrials.gov)查询,试验注册号为NCT05574296。
受试者纳入标准{14a}
纳入标准
- 入住参与研究中心的心脏重症监护室,且合并心脏疾病(包括先天性心脏病、心肌炎、心律失常及心脏移植术后排斥反应)的患者;
- 在重症监护室、心导管室或心脏手术室发生难治性心脏骤停,持续时间>5分钟,且正在接受持续心肺复苏(CPR)的患者;
- 临床团队已决定采用ECMO进行复苏的患者。注:ECMO插管需在末次胸外按压后20分钟内完成,方可判定为符合ECPR治疗标准。
排除标准
- 已加入“退出试验(opt-out)”计划的患者;
- 已知处于妊娠期的患者;
- 服刑人员;
- 本次住院期间已接受过ECPR治疗的患者;
- 自决定行ECPR治疗起,超过6小时窗口后才纳入试验的患者;
- 过去18个月内曾参与“ECMO患者基于指征的红细胞输注试验(TITRE)”的患者。
研究中心与干预实施者资质标准{14b}
设有儿童心脏重症监护室的机构具备参与本研究的资质。研究中心需完成相关方案认证、数据管理培训、临床人员培训,并满足机构审查委员会(IRB)的要求后,方可纳入受试者。研究中心还需配备ECMO医护团队(如呼吸治疗师、护士),以协助完成随机分组流程与氢气给药操作。
知情同意获取者{32a}
知情同意豁免(EFIC)与退出试验(opt-out)流程
鉴于ECPR治疗具有紧急性与生命保障性,本研究采用“知情同意豁免(EFIC)”的受试者纳入方式(图1)。本报告前文已详述作为知情同意豁免流程组成部分的社区咨询与公开告知流程。
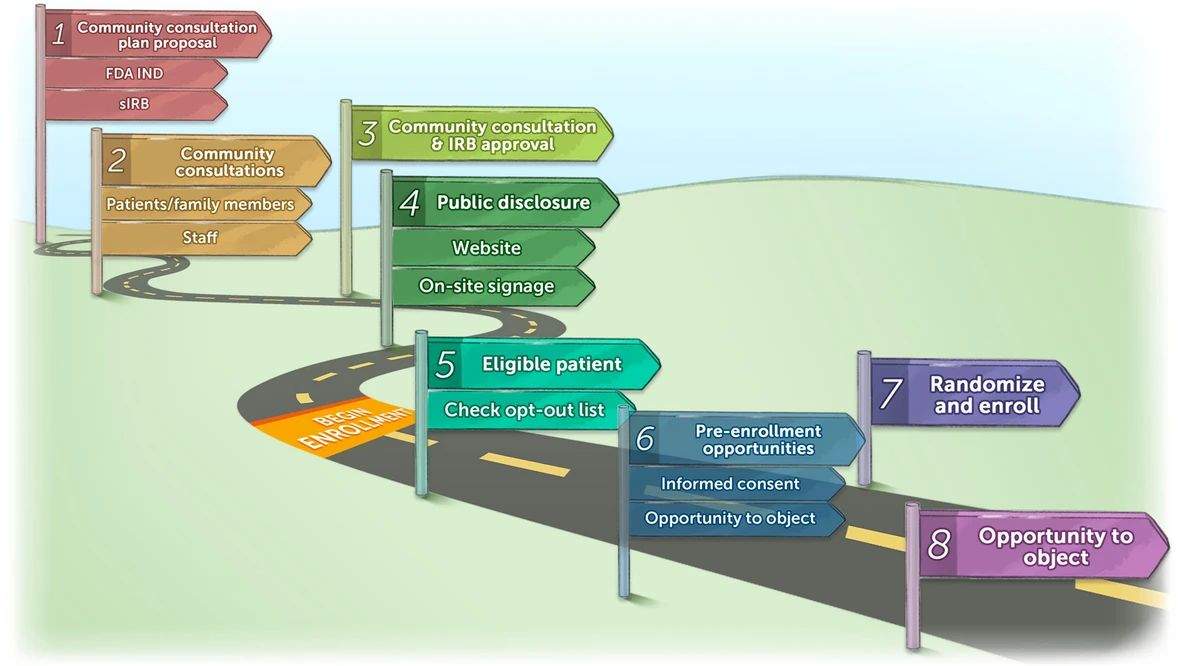
图1
采用知情同意豁免(Exception from Informed Consent, EFIC)方式开展Hydrogen-FAST试验的试验筹备及受试者纳入流程包含以下6个连续步骤:(1)社区咨询方案须同时获得美国食品药品监督管理局(FDA)及单一机构审查委员会(single IRB, sIRB)的批准;(2)初步批准后,与患者、家属及医护人员召开一系列社区咨询会议;(3)随后,收集意见与反馈并提交sIRB进行汇总审查;(4)审查通过后,进入公开告知阶段,该阶段结束后方可启动受试者纳入工作。上述1-4步骤需在每个参与研究的中心重复执行;(5)当确认符合纳入标准的患者后,医护团队成员需核查“退出试验(opt-out)名单”,确认该患者未在名单内;(6)若临床医护人员判定适宜(此类情况占少数),可向患者的法定代理人(Legally Authorized Representative, LAR)提供“入组前反对机会”,或按传统方式获取知情同意;(7)随后对患者进行随机分组并纳入试验;(8)若临床团队判定适宜,研究团队成员需向患者的法定代理人提供“反对继续参与试验的机会”。
退出试验(opt-out)流程
了解本试验的个人可通过以下方式申请退出试验:拨打各研究中心专属联系电话、发送专属邮箱,或填写各中心专属的在线表格(表格链接通过每张信息传单上的二维码获取)。若收到退出试验申请,患者信息将被加入“不予纳入名单(Do Not Enroll List)”,该名单将张贴于氢气罐及随机分组信封存放处(见图2)。已申请退出试验的住院患者还将通过佩戴“退出试验实体手环”及在病房门上张贴标识的方式进行识别。所有退出试验的识别记录需至少每日核查并确认一致性。若不同退出试验识别方式之间存在矛盾,医护人员需接受培训,优先以是否佩戴“禁止氢气治疗(No hydrogen)手环”作为判断依据。在将患者纳入试验前,负责纳入操作的医护人员需确认患者未出现在“退出试验名单”中,且未佩戴“退出试验手环”;此类患者将被排除在随机分组之外。
图2
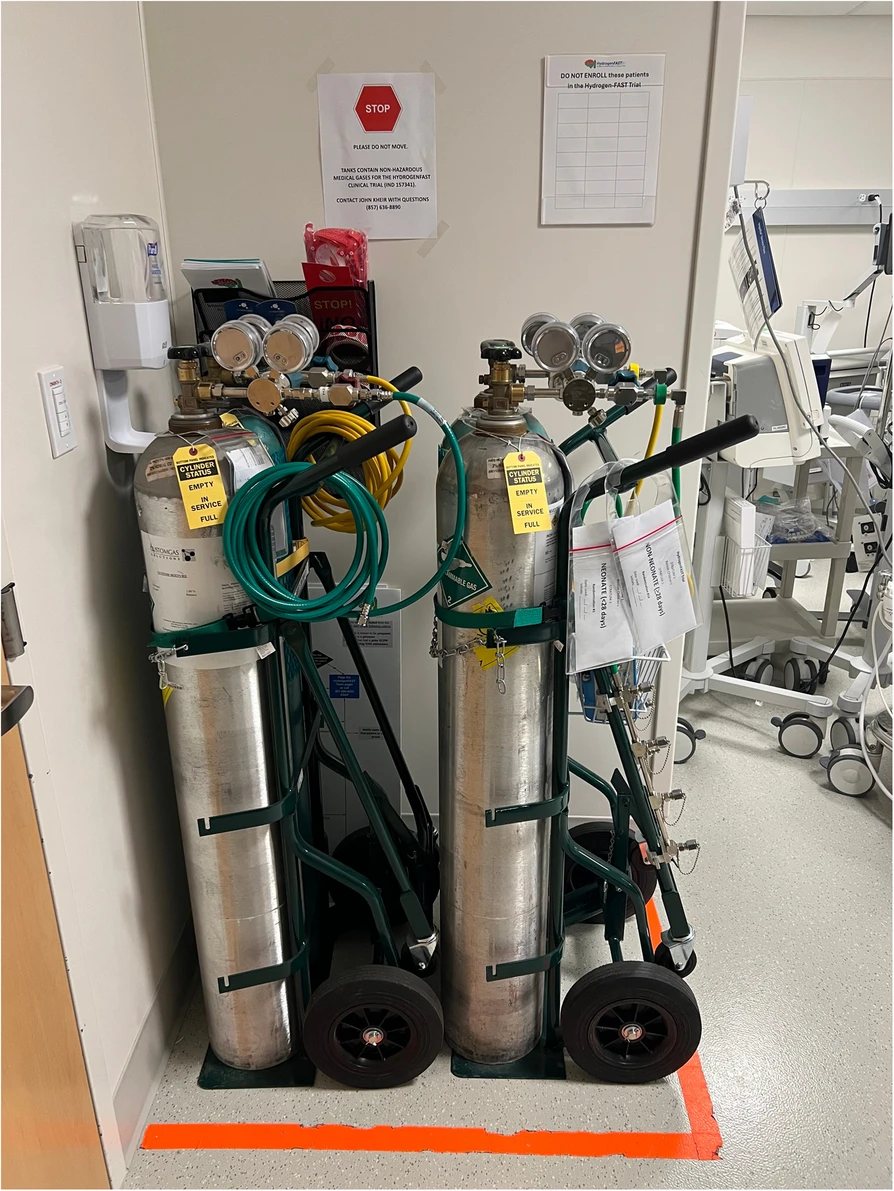
研究相关流程与方案说明
研究用产品储存与“禁止入组”清单
研究用产品需储存在安全的集中存放点。存放点相邻墙壁上需张贴“禁止入组”(Do Not Enroll)清单,清单上需为每位已选择退出研究或明确符合排除标准的患者粘贴专属标识贴。
氢气罐分为两组(一组为主罐,含汇流排;一组为备用罐),每组均固定在推车上,以便于运输。主罐推车中需放置随机化信封,供医护团队成员在现场进行随机分组操作。
反对机会
若临床条件允许,应在患者入组前为法定授权代理人(Legally Authorized Representatives, LARs)提供表达反对的机会;在极少数情况下,可采用传统知情同意流程。患者入组后,待病情稳定,需通知家属并为其提供对患者继续参与研究表达反对的机会。
表达反对的沟通应尽可能以面对面形式进行,必要时也可通过电话沟通。若家属或代理人提出反对,需立即停止对该患者(若为氢气治疗组患者)的研究干预措施,并将其退出研究。
受试者数据及生物样本收集与使用的额外知情同意规定{32b}
自患者入院起至入组后30天内,需收集患者的废弃样本并进行冷冻保存。本研究中,这些样本将用于检测脑生物标志物(包括胶质纤维酸性蛋白GFAP、tau蛋白、神经丝轻链蛋白Neurofilament light)。
在上述“表达反对的沟通”环节中,需告知法定授权代理人:本研究包含数据收集及血液样本收集。代理人可选择拒绝收集其子女的血液样本,且该决定不影响患者继续参与研究的其他部分。
干预措施与对照措施
干预措施与对照措施描述{15a}
随机分配至氢气治疗组的受试者,需通过经批准的机械通气设备及体外膜肺氧合(ECMO)回路,持续72小时吸入含2%氢气的气体。在决定为患者实施ECMO插管后,应尽快开始氢气吸入治疗。通过通气设备给予氢气的具体方法已在先前的研究中发表[9]。
研究用产品管理
氢气混合气体(含2%氢气的空气、氧气、二氧化碳,或“90/10”比例的二氧化碳与氮气混合气体)需由商业气体供应商(美国宾夕法尼亚州普拉姆斯特德维尔市斯科特医疗产品公司,Scott Medical Products, Plumsteadville, PA)提供,且需为符合药品生产质量管理规范(Good Manufacturing Practice, GMP)的认证混合气体,储存于铝制气瓶中,并通过研究专属的受控追溯系统进行管理。
每个气瓶均需附带一份分析报告(Certificate of Analysis, CoA),该报告需通过气相色谱法验证氢气浓度(可接受范围:1.95%-2.05%)。研究人员需由两人共同核对,确认气瓶序列号与分析报告一致,并验证其符合方案规定。
经确认合格的气瓶需粘贴专属研究标识,储存于上锁的限制进入区域,且该区域需与临床用气体储存区物理隔离。为确保气瓶仅用于本研究患者,需将其存放在专用位置,并配备专用调节器(该调节器与医院标准接口不兼容),且专用调节器仅由研究团队负责管理。
所有气瓶的移动与使用情况均需详细记录在追溯日志中,日志内容应包括存放位置、患者标识、气瓶识别编号及剩余压力。为确保气体供应不间断,需通过双调节器汇流排系统将气瓶串联,当其中一个气瓶耗尽时,系统会自动切换至备用气瓶。空瓶需退回供应商。
中止或修改已分配干预/对照措施的标准{15b}
在以下情况下,需在完成72小时持续氢气治疗前中止患者的氢气治疗:
- 患者本人、法定授权代理人或家属反对继续参与研究;
- 确认发生与氢气治疗存在“很可能”或“明确”关联的严重不良事件(Serious Adverse Event, SAE);
- 患者在随机分组后6小时内未接受ECMO插管;
- 患者需转运(至其他科室或机构)。
若已入组患者出现以下任一情况,需额外纳入1名新患者进入研究:
- 患者未接受ECMO治疗(即心脏骤停发生后6小时内未启动ECMO);
- 患者获知入组情况后选择退出研究。
提高干预/对照措施依从性的策略{15c}
研究中心启动入组前,需对临床工作人员进行面对面的模拟培训。每季度需开展复训,以确保工作人员持续遵守随机分组流程及已分配的随机治疗方案。
通过收集并分析氢气治疗中断及中止的报告,对工作人员遵守治疗方案的情况进行监测。
试验期间允许或禁止的合并治疗{15d}
不适用(Not applicable)。
辅助治疗与试验后治疗{34}
在体外心肺复苏(ECPR)事件发生后30天内,需每日对受试者进行监测,观察是否出现任何与氢气相关的不良事件(Adverse Event, AE)。
结局指标{12}
主要结局指标
1. 可行性:以心脏骤停后首次开始心肺复苏(CPR)为起点,计算最初72小时内氢气成功输送的时间百分比。若氢气治疗组的平均输送时间百分比≥90%,则判定氢气输送方案可行。
2. 安全性:Hydrogen-FAST试验的安全性评估核心是,在患者随机分组后30天内,对不良事件(AE)及严重不良事件(SAE)进行严格的持续监测。主要安全性结局指标为“与治疗相关”或“可能与治疗相关”的严重不良事件发生率,该发生率需由不了解患者治疗分组情况的独立专家团队判定。
不良事件的识别需通过每日查阅电子健康记录完成,并将其归入以下三个预设类别:
(1)关注的不良事件:根据研究专属分类标准明确定义并分级,包括氢气治疗相关的特定并发症(见表1);
(2)异常不良事件:即发生率、严重程度或发生时间超出体外心肺复苏(ECPR)患者群体预期的事件;
(3)非预期问题。
该分类方法可用于在“疾病相关事件高发”的高危重症患者群体中明确事件归因。氢气相关严重不良事件需按“每患者-天”计算发生率,并在不同研究组间进行比较以判定非劣效性,预设非劣效性界值为Et/Ec≤1.125(注:Et为试验组发生率,Ec为对照组发生率)。
此分类策略旨在发现可能为后续疗效试验提供参考的安全性信号,同时在复杂的临床背景下最大限度减少错误归因。
次要结局指标
表1 关注的不良事件
可行性与结局指标等相关内容
可行性
以氢气开始输送为起点,在最初连续72小时内,通过所有适用途径(如机械通气设备和体外膜肺氧合(ECMO)膜)成功输送氢气的时间百分比。
其他结局指标
- 出院存活率
- 重症监护室(ICU)住院时长与总住院时长(仅针对存活患者)
- 出院时及出院后6个月的功能状态评分(Functional Status Score, FSS)[10]
- 缺血相关实验室标志物(如pH值、乳酸、肝酶):由主要诊疗团队根据临床判断决定检测时间,数据收集周期为入组后0-30天
- 脑生物标志物(胶质纤维酸性蛋白GFAP、tau蛋白、神经丝轻链蛋白)
风险{17}
如本报告前文所述,不良事件(Adverse Event, AE)需通过每日查阅电子健康记录系统识别,并归入以下三个预设类别:(1)关注的不良事件:根据研究专属分类标准明确定义并分级,包括氢气治疗相关的特定并发症(见表1);(2)异常不良事件:即发生率、严重程度或发生时间超出体外心肺复苏(ECPR)患者群体预期的事件;(3)非预期问题。该分类方法可在“疾病相关事件高发”的高危重症患者群体中明确事件归因。
受试者时间线{18}
完整的检测时间表详见表2。自患者入院起至入组后30天内,需收集患者的废弃样本并冷冻保存。通过查阅患者记录,收集入组后30天内的结局指标数据。
表2 试验检测时间表
样本量{19}
本试验共纳入56名受试者,其中前3名为非随机化探索性受试者,均接受氢气治疗;其余53名受试者按2:1比例随机分组(32名氢气治疗组,21名对照组),以最大限度获取干预措施相关信息。
本试验的样本量计算以验证干预措施安全性为目的,基于非劣效性检验设计——即氢气治疗组的严重不良事件(SAE)发生率不劣于常规治疗组,预设非劣效性界值为12.5%。当氢气治疗组样本量为32例、常规治疗组为21例,采用单侧α=0.05检验水准,且假设两组严重不良事件发生率的相对差异为10%时,试验检验效能(power)可达85%,足以验证氢气干预措施的非劣效性。该样本量可有效识别安全性信号,为后续阶段的疗效试验提供参考。
可行性验证将采用单侧90%置信区间分析氢气平均输送时长(以心肺复苏(CPR)开始时间为起点)。当氢气治疗组纳入32名受试者时,若置信区间下限达到80%,即可证明氢气输送方案可行(平均输送时间百分比至少为90%)。
招募{20}
研究人员与临床工作人员均已接受筛选与随机分组流程培训,确保符合纳入标准的受试者能及时完成随机分组。招募工作主要通过“知情同意豁免(EFIC)”流程开展,预计通过传统知情同意流程招募的受试者占比极低。
干预措施分配:随机化
序列生成:谁生成随机序列{21a}
随机分组方案由SealedEnvelope公司生成。
序列生成:随机化类型{21b}
采用限制性随机化方法,按以下两个因素分层:(1)研究中心;(2)ECPR实施时的年龄(新生儿:出生后<28天;非新生儿:≥28天)。
分配隐藏机制{22}
随机分组方案密封于不透明信封中,信封外侧标注分层因素与信封编号。在每个分层组内,按编号顺序开启信封以确定受试者的分组方案。
实施{23}
复苏现场的医护团队成员需对符合纳入标准的患者进行随机分组评估,并前往预先指定的集中地点(见图1)——该地点张贴有清晰可见的“禁止入组”清单(由研究团队维护,列出所有已选择退出研究的患者及不符合纳入标准的患者),以提高筛选效率并快速识别非合格患者。若患者未出现在“禁止入组”清单中,负责入组的医护人员需根据研究中心与年龄分层,开启对应的实体随机分组信封。
干预措施分配:设盲
谁将设盲{24a}
不良事件评估者需对干预措施分组情况设盲。
如何实现设盲{24b}
不良事件评估者仅可查阅经编辑的不良事件描述报告,报告中不包含可识别受试者分组的信息。
必要时的破盲流程{24c}
不适用(Not applicable)。
数据收集与管理
结局指标评估与收集计划{25a}
临床数据、生理指标数据及实验室检查数据均从电子病历系统中提取;通过氢气输送日志计算氢气治疗的依从性;功能状态评分(FSS)主要通过回顾性查阅神经科与ICU病程记录确定,若记录信息不足,可通过访谈医护团队或法定授权代理人(LAR)补充。所有数据均录入符合《联邦法规汇编》第21篇第11部分(21 CFR Part 11)要求的OpenClinica数据库。
提高受试者留存率与随访完成率的计划{25b}
预计随访不完整的情况极少发生:随访期间大部分时间受试者均处于住院状态,多数数据可从电子病历系统获取;ECPR事件后6个月的最终功能状态评分(FSS)可通过多种方式完成,包括查阅电子病历、访谈医护团队或法定授权代理人。
数据管理{26}
在OpenClinica数据库中预设数据验证检查与数值范围限制,可自动生成数据质疑;此外,试验数据管理员需实时审核已录入数据,并手动发起额外的数据质疑。已制定逐条数据录入指南,指导各研究中心规范录入数据,确保数据一致性。数据导出至OpenClinica后,将存储在医院托管的密码保护服务器中,仅授权研究团队成员可访问。
保密性{33}
OpenClinica数据库会为每位筛选对象自动分配唯一的受试者ID;各研究中心需留存“受试者ID与患者身份标识”的对应关系,且仅向OpenClinica提交不含患者身份标识(日期除外)的数据。此外,已对各研究中心开展培训,指导其在向OpenClinica提交源文件时,隐去患者身份标识信息。
统计学方法
主要与次要结局指标的统计学方法{27a}
主要结局指标
对于可行性结局指标:计算氢气治疗组每位受试者在最初72小时内,通过通气设备与ECMO成功输送氢气的时间百分比;报告该百分比的均值及其95%置信区间;当组内均值≥90%时,判定氢气输送方案可行。
安全性与次要结局指标的统计学方法等相关内容
安全性结局指标
对于安全性结局指标,将计算每位受试者在入组后前30天内与治疗相关的严重不良事件(SAE)发生率。采用泊松回归模型或负二项回归模型估算事件发生率并进行组间比较,同时调整潜在混杂因素。不良事件(AE)与治疗的相关性由不良事件评估者判定。
次要结局指标
- 重症监护室(ICU)住院时长与总住院时长:主要分析采用两样本t检验(对住院时长进行对数转换后)比较组间差异,同时采用对数秩检验(logrank test)比较两组患者的出院时间分布。
- 住院期间死亡率:采用Fisher精确检验比较两组死亡率,采用对数秩检验比较两组患者的死亡时间差异。
- 功能状态评分(FSS):采用Wilcoxon秩和检验比较两组患者的功能状态评分。
- 实验室指标与生物标志物数据(缺血损伤相关实验室标志物、胶质纤维酸性蛋白GFAP、S100B蛋白、神经颗粒素):绘制指标随时间变化的趋势图;采用纵向混合效应模型(longitudinal mixed-effects models)评估组间随时间的差异,同时考虑重复测量数据的相关性。
各分析纳入的受试者范围{27b}
本1期研究不采用意向性治疗(intention-to-treat)分析方法。主要结局指标分析将纳入“随机分组后72小时内未退出研究”的随机化部分受试者。这种符合方案集(per-protocol)分析可确保纳入的患者均充分暴露于氢气治疗,从而使数据能反映氢气暴露的实际效应,而非仅反映治疗分配情况。主要及次要结局指标的次要分析方法已在试验方案中详细说明。
分析中缺失数据的处理方法{27c}
预计缺失数据量极少。系列实验室检查数据可能在部分时间点存在缺失,但本研究不计划采用数据插补(imputation)方法处理缺失值。
额外分析方法(如亚组分析){27d}
将按以下因素进行描述性亚组分析:心脏骤停发生地点(ICU、心导管室、手术室)、年龄分组(新生儿、非新生儿)、心肺复苏(CPR)持续时间。但由于检验效能有限,无法通过“治疗组与亚组的交互作用检验”检测亚组内治疗效应的差异。
中期分析{28b}
对于本早期阶段研究,目前未预设因安全性、有效性或无效性而提前终止试验的正式停止规则。试验是否终止由数据安全监察委员会(DSMB)决定,委员会将根据DSMB报告中的全部证据提出建议。
试验方案与统计分析计划{31c}
目前暂不提供统计分析计划的查阅权限。如需查阅完整试验方案,可向Kheir博士与Sleeper博士提出合理申请,审核通过后可获取。
监督与监测
协调中心与试验指导委员会的组成{3d}
Hydrogen-FAST试验由两个协调中心牵头:
- 临床协调中心:包含2名儿科心脏ICU医师研究者与1名研究助理;
- 数据与统计协调中心:包含1名博士级统计学家研究者、1名项目经理与1名数据管理员。
数据监察委员会的组成、职责与报告机制{28a}
试验由独立的数据安全监察委员会(DSMB)监督。该委员会由5名成员组成:2名儿科重症监护专家、1名儿科肾病学家兼生物伦理学家、1名生物统计学家、1名心脏外科医生。DSMB会议每半年召开一次,其核心职责包括:评估试验进展、安全性数据及关键有效性结局指标,向Hydrogen-FAST试验领导小组提供建议。
试验执行情况的审计频率与计划{29}
各研究中心将由试验团队外部的监查员进行审计。监查采用远程方式,首次监查在各中心完成首例受试者入组后开展,之后每6个月监查一次。
试验方案修订{31}
试验方案修订由两个协调中心联合实施。新修订方案起草完成后,将立即提交至单一机构审查委员会(sIRB)审批。sIRB批准新修订方案后,将向各研究中心分发新版方案,并附带一份“操作备忘录”,详细说明与上一版本相比的变更内容。所有版本的试验方案及操作备忘录均会存档于试验管理网站,供各研究中心查阅。
重大方案修订需立即提交至美国食品药品监督管理局(FDA)审查;非重大修订则在年度审查时统一提交。
研究结果传播政策{8}
研究结果将通过同行评审期刊发表、学术会议报告及公开总结报告等形式传播。
讨论
Hydrogen-FAST试验是将“氢气保护作用”的潜在临床前研究成果转化至体外心肺复苏(ECPR)患者临床应用的关键一步。缺血再灌注损伤(IRI)仍是导致儿科心脏ICU患者发病与死亡的重要原因,尤其是对于合并先天性心脏病(CHD)的患者——这类患者因复杂的心血管生理特点,本身脆弱性更高。尽管体外膜肺氧合(ECMO)技术与复苏科学不断发展,但神经损伤仍是ECPR后患者死亡或残疾的主要原因,且目前可减轻再灌注相关损伤的治疗方案十分有限[11]。
氢气之所以成为治疗缺血再灌注损伤的潜在方案,其生物学合理性在于:氢气可选择性清除细胞毒性氧自由基(尤其是羟自由基),且不会影响参与细胞信号传导的有益活性氧[12]。在心脏骤停、心肌梗死、脑卒中、器官移植等多种临床前模型中,氢气已展现出良好的药代动力学特性、扩散能力及极低的毒性[4,7,13,14,15]。值得注意的是,在一项“模拟ECPR生理状态的猪循环骤停模型”中,给予氢气治疗可改善动物的神经行为结局、减少磁共振成像(MRI)显示的脑损伤,且缺血损伤的组织学证据也显著减少[5]。
在转化应用方面,氢气在危重症患者中的应用仍处于早期阶段。包括HYBRID-II试验在内的一系列1/2期研究表明,对心脏骤停后昏迷患者给予氢气治疗具有安全性,且可能存在神经保护作用[8,16]——该研究还介绍了一种针对机械通气成人患者的氢气输送技术。而Hydrogen-FAST试验采用的氢气输送方法具有独特性:通过“经批准但未改装”的ICU通气设备,输送预先认证的氢气混合气体[9]。这种方法允许自由调整吸入氧浓度,且支持患者自主呼吸,克服了此前氢气输送方法的两项主要限制。
Hydrogen-FAST试验的另一独特之处在于,将这种新干预措施整合到“儿科ECPR”这一高度紧急且伦理复杂的场景中。由于ECPR具有时间紧迫性,几乎无法获取传统知情同意,因此本研究采用“知情同意豁免(EFIC)”机制纳入受试者。在复苏研究中,知情同意豁免已被越来越多地认可为必要且合理的监管工具,但前提是需实施完善的社区咨询与退出试验(opt-out)流程[17,18]。Hydrogen-FAST试验严格遵循美国FDA《联邦法规汇编》第21篇第50.24节(21 CFR 50.24)的规定,通过社区咨询、公开告知及“入组前后反对机会”等多层利益相关者参与机制,构建了一套可被其他“紧急场景下儿科试验”借鉴的框架。
本研究特意设计为1期可行性与安全性试验。通过重点收集“方案依从性、严重不良事件发生率、生物标志物变化趋势”等严谨数据,Hydrogen-FAST试验将为“是否推进至更大规模疗效试验”提供关键初步证据。尽管该样本量不足以检测神经结局或生存率的微小差异,但足以识别可行性障碍与重大安全性问题。此外,本研究收集的储存血清样本与神经影像学数据,将为探索性结局指标分析提供支持,进而为未来2/3期试验的设计提供参考。
若试验取得成功,其意义将超越儿科ECPR领域。氢气治疗或可在其他以缺血再灌注损伤为特征的危重症中探索应用,包括院外心脏骤停、新生儿缺氧缺血性脑病、体外循环、实体器官移植等。此外,为氢气输送开发的基础设施(如气体输送系统、监测工具、给药方案)可在各机构推广,助力氢气治疗的广泛临床应用。
Hydrogen-FAST试验在创新监管框架下,采用机制靶向性干预措施,解决了迫切的临床需求。试验通过标准医疗设备输送2%氢气,且使用经生产认证的混合气体,降低了氢气在临床环境中应用的风险。这项随机试验的结果不仅将深化我们对氢气治疗的理解,还将为儿科危重症研究方法的发展做出贡献。
试验状态
- 试验方案版本:2.5版(2025年7月21日)
- 新药临床试验申请(IND)编号:157341(2023年6月激活)
- 临床试验注册平台(ClinicalTrials.gov)编号:NCT05574296
- 招募启动时间:2024年3月
- 预计研究完成时间:2027年8月31日
https://blog.sciencenet.cn/blog-41174-1509781.html
上一篇:首个慢性疲劳综合征血液检测方法问世
下一篇:嗜热菌氢气的代谢调节作用
