博文
活性氧导致细胞微核崩溃《科学》封面  精选
精选
|
高频率的染色体错误分离,被称为染色体不稳定性,是肿瘤的一个普遍特征。错误分离的染色体可以被封装到微核中,微核是一种异常的细胞核结构,当其包膜破裂时,会促进复杂染色体重排的获得。两组研究人员现在提供了关于微核崩溃及其对癌症过程的影响的机制性见解(参见Maddaluno和Settembre的观点文章)。Martin等人报告说,线粒体衍生的活性氧(ROS)氧化自噬蛋白p62,增强其对修复机械组件的自噬活性,从而增加微核崩溃。在癌细胞和高染色体不稳定性肿瘤中,p62水平的升高与染色体重排的增加有关,这表明p62可能是这类肿瘤的潜在预后标志物。Di Bona等人发现,ROS干扰一种名为CHMP7的膜修复蛋白,该蛋白通常有助于维持核膜的完整性。ROS导致CHMP7聚集并与另一种蛋白LEMD2结合,导致微核破裂。这种崩溃促进了遗传异常和炎症,将氧化损伤与癌症的侵袭性行为联系起来。

引言
染色体不稳定性是侵袭性癌症的标志之一,其特点是存在微核,这些是包含整条染色体或染色体臂的、易于破裂的胞质结构。微核包膜的不可逆崩溃是肿瘤进展中的一个中心事件。微核崩溃使封装的DNA暴露于胞质中,催化染色体重排和表观遗传改变,这些改变驱动了肿瘤异质性以及治疗抵抗。微核破裂还激活了炎症信号传导途径,重塑了肿瘤免疫微环境,促进了转移。尽管其重要性,但微核崩溃的底层机制尚不清楚。
推理
维持核膜的完整性对于细胞功能和生物体的存活至关重要。尚不清楚在微核中哪些细胞防护措施受损,导致核膜完整性的破坏。我们推测,微核与主核之间的内在差异可能解释了它们在膜稳定性上的不同。例如,微核比主核小5到20倍。其次,它们拥有异常的核膜,如异常的核孔功能和减少的lamin B1水平所示。此外,一旦破裂,微核包膜很少被修复。
结果
在这项研究中,我们将先前的观察与我们自己的发现结合起来,即微核包膜破裂是由于微核与线粒体之间的异常相互作用所致,这一过程由活性氧(ROS)介导。实际上,与完整的微核相比,破裂的微核更有可能与线粒体网络混合在一起,而调节ROS水平相应地改变了微核破裂的比率。随后的蛋白质组学分析和遗传操作使我们观察到,ROS通过干扰称为运输所需的内体分选复合物III(ESCRT-III)的核膜修复复合物的正常活动来干扰微核的完整性。ROS水平的增加导致ESCRT-III支架蛋白带电多囊体蛋白7(CHMP7)在微核中的积累,促进了非典型功能。ROS抑制了CHMP7的输出,导致其在微核中持续存在并与内核膜伴侣LEM结构域核膜蛋白2(LEMD2)异常结合。ROS诱导的半胱氨酸氧化催化了CHMP7聚集并减少了其与ESCRT-III复合物中的规范结合伴侣的相互作用。CHMP7聚集体与LEMD2的结合促进了微核膜变形和崩溃。这种效应进一步被ROS依赖性招募自噬相关蛋白p62所加剧,后者介导了规范ESCRT-III成员的降解,限制了随后膜修复的任何机会。最后,我们发现这一机制与人类肿瘤相关。ROS水平的升高和CHMP7功能的异常导致了复杂的染色体重排,或称为染色体粉碎现象,已知这是由微核破裂引起的。此外,缺氧条件下产生的ROS以CHMP7依赖的方式诱导微核破裂。与这些发现一致,我们在人类头颈癌和卵巢肿瘤的缺氧区域观察到微核破裂和CHMP7积累的显著增加。
结论
我们的结果揭示了微核与线粒体之间的病理相互作用,这是微核破裂的基础。通过指出ROS作为微核完整性的调节因子,这项工作为ROS诱导条件(如自由基生成和缺氧)与已知由微核存在引起的下游过程(包括染色体重排、表观遗传失调和促进肿瘤的炎症)之间提供了机制性联系。

微核破裂的机制:
线粒体与微核的接近通过线粒体衍生的活性氧物种(ROS)驱动微核膜破裂。ROS抑制微核输出,导致CHMP7(一种与核膜修复复合体ESCRT-III相关的支架蛋白)的过度积累。ROS依赖的半胱氨酸氧化促进CHMP7自我聚集并异常结合到膜蛋白LEMD2上,导致微核崩溃。
摘要:
含有染色体的微核是侵袭性癌症的标志。微核经常发生不可逆的崩溃,将其封闭的染色质暴露于胞质中。微核破裂催化染色体重排、表观遗传异常和炎症,但保护微核完整性的机制尚不清楚。在这项研究中,我们发现线粒体衍生的活性氧物种(ROS)通过促进带电多泡体蛋白7(CHMP7)的非规范功能来破坏微核,CHMP7是一种膜修复复合体,即内体分选复合物所需运输III(ESCRT-III)的支架蛋白。ROS将CHMP7保留在微核中,同时破坏其与其他ESCRT-III组分的相互作用。ROS诱导的半胱氨酸氧化刺激了CHMP7的寡聚化并结合到核膜蛋白LEMD2上,破坏了微核包膜。此外,这一ROS-CHMP7病理轴引起了已知由微核破裂引起的染色体粉碎。它还在缺氧条件下介导了微核解体,将肿瘤缺氧与推动癌症进展的下游过程联系起来。
哺乳动物细胞已经进化出健壮的机制来保护核膜的完整性。然而,多种生理和病理过程导致核膜破裂,需要迅速修复。内体分选复合物所需运输III(ESCRT-III)复合体是介导核膜修复的关键因素,它通过与LEM结构域核膜蛋白2(LEMD2)和带电多泡体蛋白7(CHMP7)支架在核膜破裂位点的结合而组装。尽管对主核的膜修复进行了广泛研究,但对于调节含染色体微核完整性的机制了解较少。微核是具有染色体不稳定性(CIN)的癌细胞的标志,这一特征与预后不良、治疗抵抗和远处转移有关。
微核是由有丝分裂期间染色体分离错误产生的,其特点是具有异常的核膜和减少的lamin B含量。与主核不同,微核包膜破裂通常是不可逆的,因此将封闭的染色质暴露于胞质中。已显示微核破裂能催化复杂的基因组重排,如染色体粉碎,以及长期的表观遗传异常,这些异常促进了肿瘤的进化。此外,破裂的微核是胞质双链DNA的主要来源,激活了环状GMP-AMP合成酶和干扰素基因刺激子(cGAS-STING)先天免疫途径。在正常情况下,cGAS-STING激活介导促炎信号传导,可以发挥抗肿瘤活性;然而,在染色体不稳定的肿瘤细胞中,这一途径的慢性激活也被显示能促进肿瘤促进性炎症,导致转移进展。微核崩溃的不可逆性引发了与主核不同的膜完整性和修复机制的内在缺陷。最近的研究已经确定了内质网膜侵入微核膜、核孔复合体形成缺陷以及微核层减弱作为微核与主核区别的特征。然而,关于微核破裂和修复缺陷的确切机制仍不清楚。
线粒体衍生的ROS促进了微核崩溃
在HeLa细胞中,我们观察到微核与线粒体之间显著的空间重叠。此外,与远离线粒体网络的微核相比,与线粒体网络进行广泛且长时间接触的微核更有可能破裂。对主核和微核进行的无偏见蛋白质组学分析揭示了微核中富集的线粒体相关蛋白。我们假设,接近线粒体会使微核暴露于活性氧物种(ROS),影响微核包膜的完整性。电子从电子传递链(ETC)的复合物I和III泄漏构成了线粒体衍生ROS的主要来源。为了测试ROS在破裂中的作用,我们在一组七个细胞系中评估了加入过氧化氢(H2O2)或用 oligomycin 或 VLX600处理后的微核完整性,这些物质是已知的线粒体ETC破坏剂。使用多种正交方法评估微核完整性。存在lamin B1、lamin A(20)、在赖氨酸9处乙酰化的组蛋白H3(H3K9Ac)或融合到核定位序列(NLS)的红色荧光蛋白(RFP)表示完整的微核。相反,它们的缺失或cGAS(28)的存在,后者在失去微核膜完整性后会迅速被招募,表明微核破裂。每种处理条件下的ROS水平通过基于发光的测定法在细胞裂解物中测量。直接添加H2O2或破坏线粒体ETC导致破裂微核的比例显著增加,而对微核总数几乎没有影响。相反,用N-乙酰半胱氨酸(NAC)、线粒体ROS清除剂MitoQuinone (MitoQ) 或过氧化氢酶处理甲状腺永生化NTHY Ori 3.1(NTHY)、骨肉瘤(U2OS)或宫颈癌(HeLa)细胞一致减少了微核破裂的发生率。虽然破裂的微核更有可能靠近线粒体,但对过氧化物酶体来说并非如此,过氧化物酶体也是细胞ROS的另一个来源。过氧化氢酶限制了过氧化物酶体外的ROS泄漏。因此,通过添加脂肪酸诱导过氧化物酶体ROS并没有在有过氧化氢酶存在下增强微核破裂,而通过抑制酰基辅酶A氧化酶1(ACOX1)抑制过氧化物酶体ROS的产生也没有减少破裂。因此,微核破裂的机制涉及线粒体衍生的ROS,特别是那些影响微核包膜完整性的ROS。因此,线粒体来源的ROS被证明是微核破裂的一个重要贡献者。

图1. 线粒体ROS是微核破裂的原因。
(A)HeLa细胞的典型荧光(“fluo”,顶部)和重建的3D(“rec”,底部)图像;黄色表示核完整性标记物(NLS-RFP),洋红色表示线粒体,蓝色表示DNA(Hoechst 33342)。比例尺,6 μm。MN,微核。(B)来自三个生物重复的六十张图像(A)用Imaris分析。线粒体与完整(NLS-RFP+)和破裂(NLS-RFP−)的微核(MNi)和主核(PN)的相对体积重叠。非配对Student’s t检验,Welch校正。(C)HeLa细胞的典型图像;黄色表示NLS-RFP,蓝色表示H2B-iRFP,绿色表示线粒体。比例尺,3 μm。(D)来自三个生物重复的总共三十个时间序列(C)如(B)中分析。完整(灰色)和破裂(红色)MNi-线粒体的相对体积重叠随时间变化(MVO%,最大体积重叠百分比)。对于完整MNi,时间0表示成像结束。平均值±SEM。(E)(D)中的MNi根据MVO%的中位数分为两个群体。Log-rank(Mantel-Cox)检验(P = 0.0093)和Gehan-Breslow-Wilcoxon检验(P = 0.0095)。(F)富含破裂(cGAS-GFP+)的MNi蛋白质与MDA-MB-231细胞的主核(PN)蛋白质相比(见材料和方法及表S1)。KEGG(京都基因与基因组百科全书)途径分析MNi富集蛋白(洋红色表示线粒体特异性过程;绿色表示不仅限于线粒体涉及的过程)。(G)NTHY细胞中完整(顶部,lamin B+)和破裂(底部,lamin B−)的MNi。比例尺,3 μm;插图,1 μm。(H)VLX600和鱼藤酮的作用靶点。(I至N)经过ROS调节后破裂MNi(G)的量化。最少三个生物重复,平均值±SD,非配对Student’s t检验。f.c.表示平均倍数变化的平均值。(I)经过24小时VLX600处理后的破裂MNi;n = 300 MNi。(J)经过4小时鱼藤酮处理后的破裂MNi;n = 300 MNi。P值:NTHY,0.0271;U2OS,0.0388;HeLa,0.0047。(K)经过4小时H2O2处理后的破裂MNi;n = 100 MNi。P值:NTHY,0.0002;U2OS,0.0229;HeLa,0.0124。(L)经过24小时NAC处理后的破裂MNi;n = 100 MNi。P值:NTHY,0.0490;U2OS,0.0029;HeLa,0.0104。(M)经过24小时MitoQ(mitoquinone mesylate)处理后的破裂MNi;n = 100 MNi。P值:NTHY,0.0002;U2OS,0.0094;HeLa,0.0152。(N)经过4小时过氧化氢酶处理后的破裂MNi;n = 100 MNi。P值:NTHY,0.0036;U2OS,0.0006;HeLa,0.0009。
通过ESCRT-III非依赖性CHMP7功能导致微核破裂
然后我们询问了ROS是否破坏了已知定位于微核的ESCRT-III核膜修复复合体的功能(图2A)。先前的报告已经暗示,ESCRT-III功能的过度活跃是诱导微核破裂的潜在机制(21)。我们生成了稳定表达多西环素可诱导Cas9和针对CHMP7、该复合体的支架蛋白以及带电多泡体蛋白2a(CHMP2A)和4b(CHMP4B),两个必需的ESCRT-III亚基的单向导RNA的HeLa细胞(图S5A)(37)。可诱导敲除(KO)CHMP7完全消除了ROS诱导的微核破裂(图2B)。然而,敲除CHMP2A或CHMP4B对这一过程没有影响(图2B和图S5,B至M)。此外,在未处理或载体处理的条件下,单独消耗CHMP7减少了破裂微核的基础发生率(图S6,A至D)。而且,用NAC处理CHMP7-KO细胞并没有进一步减少微核破裂(图2C和图S6,E和F)。组成型表达抗CRISPR的[在原型空间序列相邻基序(PAM)序列中带有无义突变,以下简称PAM*] CHMP7在CHMP7-KO细胞中完全恢复了ROS诱导的微核破裂,以及对微核的保护作用(图2, D和E)。因此,我们发现CHMP7蛋白介导了独立于其规范ESCRT-III功能的ROS诱导的微核破裂。
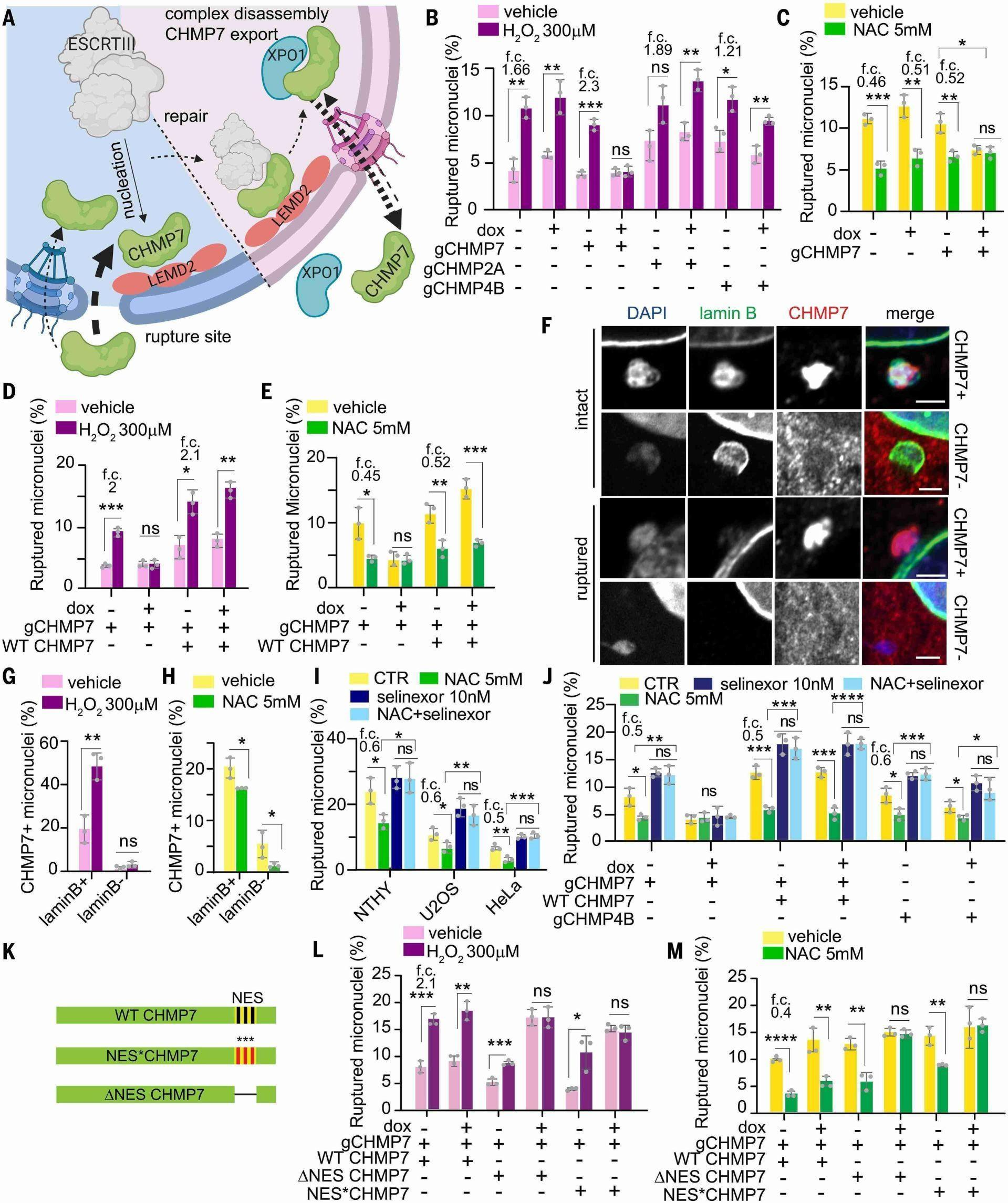
图2. ROS通过ESCRT-III非依赖性的CHMP7作用导致破裂。
(A) 在核内扩散的CHMP7被XPO1导出。当主核破裂时(左),CHMP7饱和XPO1并结合其伴侣LEMD2。这启动了ESCRT-III复合体的组装,修复了孔洞(右),然后XPO1将所有这些分子导出。(B至E) 在表达多西环素(dox,1 μg/ml,处理72小时)诱导型Cas9(iCas9)和指示的向导RNA(gRNA),以及带有或不带有PAM* CHMP7野生型(WT),用H2O2或NAC处理的HeLa中破裂的微核。(B) H2O对比H2O2的P值:WTdox−, 0.0024; WTdox, 0.005; gCHMP7dox−, 0.001; gCHMP2Adox, 0.0036; gCHMP4Bdox−, 0.0122; gCHMP4Bdox, 0.0034。(C) H2O对比NAC的P值:WTdox−, 0.0006; WTdox, 0.0031; gCHMP7dox−, 0.0066; gCHMP7H2O dox−对比dox, 0.0133。(D) H2O对比H2O2的P值:gCHMP7dox−, 0.0026; CHMP7 WTdox−, 0.0109; CHMP7 WTdox, 0.0011。(E) H2O对比NAC的P值:gCHMP7dox−, 0.0188; CHMP7 WTdox−, 0.0086; CHMP7 WTdox, 0.0009。(F) HeLa中CHMP7+和CHMP7−完整和破裂的微核。比例尺,2 μm。(G和H) (F)中用H2O2或NAC处理的CHMP7+MNi。(G) P = 0.0053。(H) P值:lamin B+, 0.0463; lamin B−, 0.0442。(I) 用selinexor、NAC或两者处理24小时的破裂微核。NTHY的P值:对照(CTR)对比NAC,0.0232; NAC对比两者,0.0101。U2OS的P值:CTR对比NAC,0.0359; NAC对比两者,0.0074。HeLa的P值:CTR对比NAC,0.0021; NAC对比两者,<0.0001。(J) 如(B)至(E)所述在HeLa中处理如(I)的破裂微核。P值:gCHMP7dox− CTR对比NAC,0.0172; gCHMP7dox− NAC对比两者,0.0014; CHMP7 WTdox− CTR对比NAC,0.0008; CHMP7 WTdox− NAC对比两者,0.0007; CHMP7 WTdox CTR对比NAC,0.0006; CHMP7 WTdox NAC对比两者,<0.0001; gCHMP4Bdox− CTR对比NAC,0.0226; gCHMP4Bdox− NAC对比两者,0.001; gCHMP4Bdox CTR对比NAC,0.0279; gCHMP4Bdox NAC对比两者,0.0172。(K) 出口缺陷型CHMP7突变体。(L和M) 在HeLa CHMP7 KO中用(K)处理并用H2O2或NAC处理的破裂微核。(L) H2O对比H2O2的P值:WTdox−, 0.0004; WTdox, 0.0013; ΔNESdox−, 0.0008; NES*dox−, 0.0193。(M) H2O对比NAC的P值:WTdox−, 0.0019; WTdox, 0.0005; ΔNESdox−, 0.0039; NES*dox−, 0.0049。对于(B)至(M),n = 100 MNi,三个生物重复,平均值±SD,非配对Student’s t检验。
ROS促进微核中CHMP7的积累
通过高分辨率成像观察表达CHMP7-绿色荧光蛋白(GFP)的HeLa细胞,我们发现ROS主要促进CHMP7在lamin B阳性(lamin B+)(即完整)微核中的积累(图2,F和G)。通过染色内源性CHMP7在甲状腺癌细胞中获得相似的结果(图S6,G至J)。通过活细胞成像证实这一观察结果,其中CHMP7定位到微核先于微核区室化的丧失(图S7)。相反,用NAC处理减少了微核中CHMP7的积累(图2H)。CHMP7从核中的导出依赖于核输出受体exportin XPO1,后者识别CHMP7上的核输出序列(NES)(5)。ROS可以通过O-连接的N-乙酰葡糖胺酰化(O-Glc-NACylation)修饰核孔蛋白(Nup),从而增加它们与XPO1的结合并限制其移动性,这对它的输出功能至关重要(38)。这促使我们询问ROS是否通过抑制XPO1依赖性输出来破坏微核中CHMP7的输出。实际上,诱导ROS增加了核膜上的O-Glc-NAC信号(图S8,A至C)。然后我们对用H2O或H2O2处理过的纯化微核进行液相色谱-质谱(LC-MS)分析(39,40),发现在升高的ROS条件下微核中相对富集了XPO1(图S8,D和E,以及表S3)。此外,在氧化条件下瞬时表达与黄色荧光蛋白(YFP)融合的XPO1的细胞中,我们观察到在微核包膜处XPO1的积累发生在破裂前,这表明异常结合的增加干扰了其输出功能(图S8,F至J)。
接下来,我们使用泛核输出抑制剂莱普霉素B(LMB)或特异性XPO1抑制剂selinexor处理HeLa和U2OS细胞。用任一药物处理都显著增加了微核破裂的频率,消除了ROS进一步增加破裂频率的能力,并完全阻止了NAC诱导的微核包膜稳定化(图2I和图S8,K至L)。条件性敲除CHMP7——而非CHMP4B——消除了selinexor对微核破裂的影响(图2J)。接着,我们在携带条件性CHMP7敲除的细胞中表达了带有突变或截短核输出序列(NES*和ΔNES,分别为)的PAM* CHMP7(图2K)。表达任一突变体导致CHMP7在微核中组成性积累,并诱导了ROS和ESCRT-III非依赖性的微核破裂增加,消除了NAC的膜稳定作用(图2,L和M,以及图S9,A至F)。因此,我们展示了未能输出CHMP7介导了ROS依赖性的微核破裂增加。
CHMP7通过聚集并与LEMD2结合诱导破裂
鉴于ESCRT-III功能对ROS诱导的微核破裂不是必须的,我们试图了解CHMP7,一个自身没有任何已知膜重塑功能的蛋白,是如何使微核塌陷的。利用高分辨率显微术和三维(3D)重建,我们发现CHMP7+微核的特点是核纤层变形增加,这表明与纤层组分结合(图3,A和B)。为了确定微核中可能的ROS依赖性CHMP7结合伴侣,我们从用H2O2或NAC处理过的纯化微核中免疫沉淀CHMP7,并对沉淀物进行LC-MS分析(图3,C和D,以及表S4)。ROS导致了CHMP7结合的转变:在NAC条件下,CHMP7结合了ESCRT-III修复复合体的亚单位CHMP2A和CHMP4B,这一发现通过从全细胞裂解物中沉淀下来的CHMP7的免疫印迹得到确认(图3D和图S10,A和B)。相反,在升高的ROS条件下,CHMP7更有可能结合LEMD2,一种内核膜结构蛋白,这一点通过LEMD2相互沉淀得到确认(图3D和图S10,B和C)。
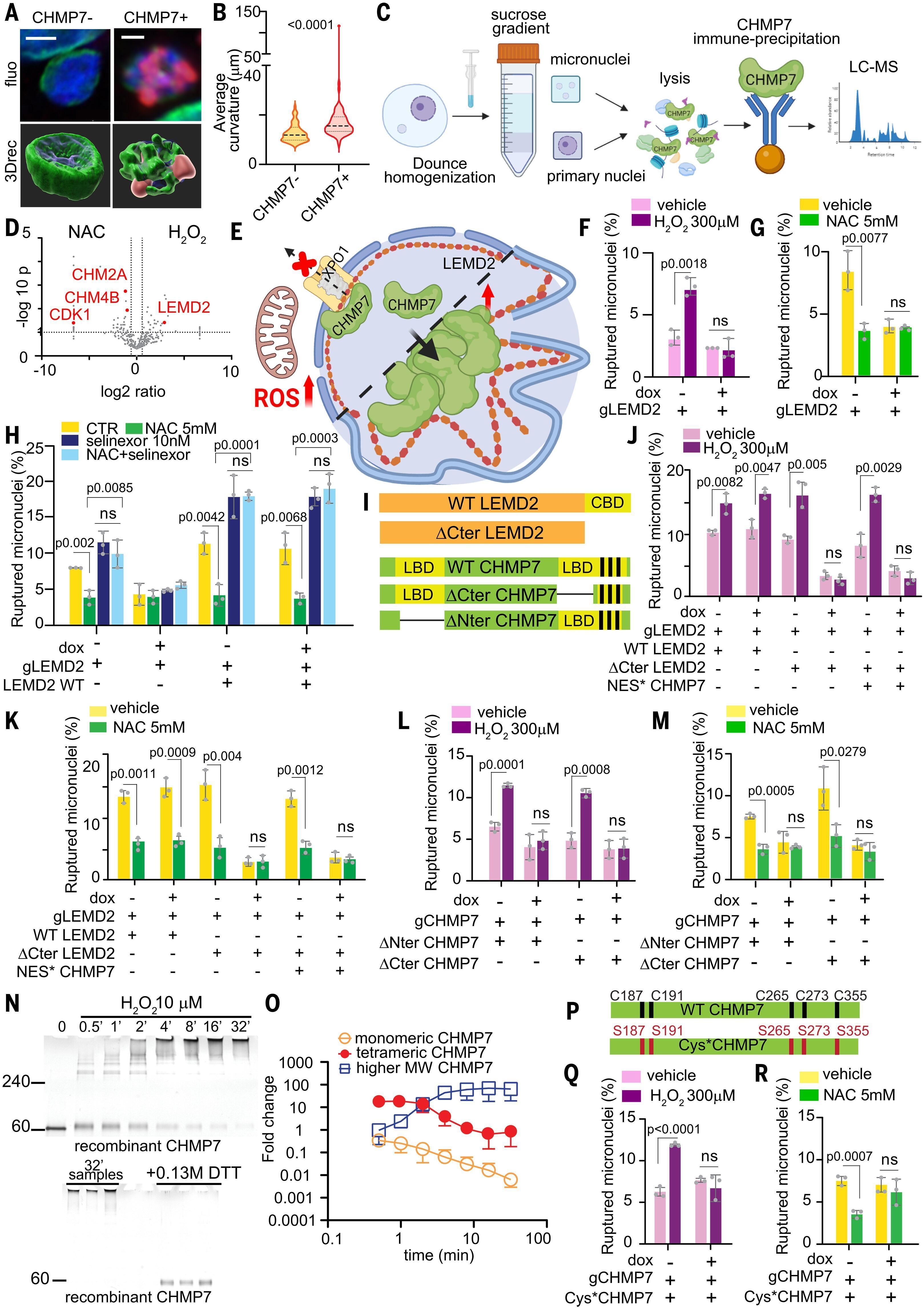
图3. CHMP7通过与LEMD2结合和聚集诱导破裂。
(A) 代表性的CHMP7(红色)+和−微核。蓝色,DAPI;绿色,lamin B1。比例尺,0.5 μm。(B) 使用Imaris(见材料和方法)对(A)中的顶点平均曲率分布进行分析。n = 15个微核,三个生物学重复,Mann-Whitney检验。(C) 通过LC-MS检测CHMP7结合伴侣的实验流程,从提取的微核中免疫沉淀CHMP7 (39)。(D) (C)的结果,三个生物学重复。(E) 线粒体ROS抑制XPO1介导的CHMP7输出,导致其在微核中积累,寡聚并结合LEMD2,拉动它和纤层直至破裂。(F 和 G) 在表达iCas9和针对LEMD2的向导RNA的HeLa中用H2O2或NAC处理的破裂微核。(H) 在有无PAM* LEMD2 WT并用selinexor、NAC或两者处理的HeLa LEMD2 KO中的破裂微核。(I) CHMP7和LEMD2结合缺陷的突变体。(J 和 K) 如(F)和(G)所示,在表达(I)中所示突变体或与ΔCter LEMD2一起过表达CHMP7 NES*,并用H2O2或NAC处理的HeLa中的破裂微核。(L 和 M) 在表达(I)中所示突变体并用H2O2或NAC处理的HeLa iCas9 gCHMP7中的破裂微核。(N) (顶部)重组CHMP7与10 μM H2O2反应指定时间后的凝胶电泳。(底部) 图像显示如(顶部)中混合了0.13 M DTT以还原二硫键的32分钟反应,显示单体CHMP7。(O) 来自如(N)进行的三个生物学重复实验的CHMP7单体、四聚体和高分子量寡聚体的定量。(P) 缺乏半胱氨酸的CHMP7突变体。(Q 和 R) 在表达(P)中所示突变体并用H2O2或NAC处理的HeLa iCas9 gCHMP7中的破裂微核。对于(F)到(R),n = 100个微核,三个生物学重复。平均值±SD,非配对Student’s t检验。
因此,我们假设ROS依赖性的CHMP7-LEMD2相互作用诱导了微核包膜的变形和塌陷(图3E和图S10D)。为了测试这一点,我们在HeLa iCas9细胞中生成了可诱导的LEMD2 KO。LEMD2的消耗保护了细胞免受ROS诱导的微核破裂,并完全恢复了微核完整性至NAC处理所见的水平(图3,F到H,以及图S10,E到G)。此外,在CHMP7 KO和LEMD2 KO条件下,微核变形减少,我们通过纯化微核的原子力显微镜图像确认了这些发现(图S10,H到J)。然后我们用PAM* LEMD2或缺少CHMP7结合域(CBD,称为ΔCter LEMD2)的截短版LEMD2来拯救可诱导的LEMD2 KO(图3I)(41, 42)。用ΔCter LEMD2补充LEMD2 KO导致了微核包膜完整性的恢复,对ROS不敏感,并且总体上减少了微核中CHMP7的保留(图3,J和K,以及图S11A)。这一结果表明,微核中CHMP7的保留部分依赖于输出抑制以及通过与LEMD2结合而浓缩。此外,在表达ΔCter LEMD2的细胞中的微核也对核输出抑制或CHMP7 NES*的过表达有抵抗力,这在微核中累积并通常会促进显著的微核包膜塌陷(图3,J和K,与图2,K到M,以及图S11B相比)。因此,我们发现微核中CHMP7的积累是诱导破裂的必要条件,但不是充分条件,需要CHMP7-LEMD2结合。相应地,用LEMD2结合域(LBD)缺陷的突变体[称为ΔCter和ΔNter CHMP7(41)]补充CHMP7 KO消除了ROS依赖性的微核破裂和膜变形(图3,L和M,以及图S11,C和D)。
CHMP7依赖的微核破裂受到细胞周期蛋白依赖性激酶1(CDK1)的抑制。
我们的CHMP7-IP分析揭示了,除了ESCRT-III组分外,NAC处理促进了CHMP7与细胞周期蛋白依赖性激酶1(CDK1)的相互作用(图3D和表S4)。已知CDK1介导的CHMP7磷酸化会影响其与LEMD2的相互作用(42)。在CHMP7上,丝氨酸3(S3)和丝氨酸441(S441)的磷酸化阻碍了其在规范的ESCRT-III功能背景下与LEMD2的结合(42)(图S12A)。为了测试ROS是否影响CHMP7的磷酸化,我们从用H2O2或NAC处理过的细胞中免疫沉淀CHMP7后进行了磷酸蛋白质组学分析。增加的ROS水平导致CHMP7-S3磷酸化显著减少(图S12,B到E)。我们注意到,与初级核相比,微核中的CDK1活性显著降低,这通过前者中视网膜母细胞瘤蛋白(pRb)染色的减少得到了证明(图S12,F到I)。然后,我们使用两种不同浓度(1 μM和10 μM)的CDK1特异性抑制剂Ro3306来评估CDK1活性对微核破裂的影响。只有高剂量的抑制导致了细胞周期停滞;然而,在1 μM剂量下,Ro3306仍然显著减少了微核中的pRb(图S12,L到O)。与此发现一致,在任何浓度下药理抑制CDK1都显著增加了微核破裂(图S13 A和B)。CDK1抑制剂的效果在用NAC处理后完全得到挽救,并且在用selinexor进行核输出抑制时进一步加剧(图S13,C和D),这表明防止CHMP7-S3和-S441磷酸化增强了其与LEMD2的结合,而不是其在微核中的积累。因此,用Ro3306处理并没有促进CHMP7在微核中的积累(图S13,E和F)。
为了补充我们的药理学发现,我们在其他情况下表达含有抗磷酸化突变体的CHMP7蛋白,在丝氨酸残基上(例如,S3A CHMP7和S441A CHMP7)或在两个残基上(P*CHMP7)在CHMP7 KO细胞中(图S13G)(在这些突变体中,我们指示了原始氨基酸残基的位置以及我们用来替换它的残基;例如,S3A表示第3位的丝氨酸被丙氨酸替换)。表达这些突变体中的任何一个都导致了微核破裂的增加,这在NAC处理后得到挽救(图S13,H和I),反映了我们的药理学发现。总的来说,这些结果支持了这样一个观点:通过ROS加剧微核破裂的次级机制是磷酸化介导的CHMP7-LEMD2结合的破坏,并且这一机制依赖于CHMP7在微核中的积累。CHMP7的磷酸蛋白质组学分析揭示了CHMP7中其他ROS依赖性的磷酸化位点(例如,S410、S417和S429),其中一些是核因子κB(NF-κB)途径中激酶的潜在目标,暗示了一个潜在的调节作用(图S12,C和D)(43)。
ROS促进了CHMP7的寡聚和半胱氨酸氧化
CHMP7+微核的物理变形让人想起了一个可能涉及CHMP7聚集的潜在机制(图3,A和E)。H2O2与硫醇(-SH)反应形成二硫键(-S-S)(图S14)(30, 44)。我们假设线粒体衍生的H2O2与CHMP7的一个或多个五个半胱氨酸残基的硫醇部分反应,形成与LEMD2相互作用的寡聚体,从而使微核变形。为了测试这一点,我们首先询问了H2O2的存在是否会干扰微核定位的CHMP7聚集体中的动态。我们对表达CHMP7-GFP的活细胞进行的荧光恢复后光漂白分析显示,当细胞暴露于H2O2时,微核CHMP7聚集体经历了较少的可扩散和结合CHMP7之间的蛋白交换,表明增强了结合强度和持久性(图S14,A到C)。
接下来,我们使用非还原凝胶电泳分析了H2O2处理对纯化的重组CHMP7高阶结构的影响。在反应中添加H2O2到单体CHMP7中导致在30秒内出现两个主要变化:形成了一个迁移速度较慢的CHMP7形式(CHMP7′)和形成了CHMP7四聚体。随着反应的进行,CHMP7′和CHMP7四聚体被消耗以形成在8分钟时主导反应的更高寡聚体(图3,N和O,以及图S14D)。这些高分子量异构体在用还原剂二硫苏糖醇(DTT)处理后消失,表明寡聚体通过分子间二硫键稳定(图3N)。同样可能的是,CHMP7′含有一个促进分子间硫醇-二硫交换形成各种寡聚体的分子内二硫键(图S14E)。这些反应是非特异性的且与序列无关,因此可以在CHMP7和具有半胱氨酸残基的其他蛋白质之间发生。因此,LEMD2中存在几个半胱氨酸残基,而CHMP2A和CHMP4B中缺乏此类残基,这可能为在ROS增加时观察到的CHMP7结合伴侣的转变提供了解释(图3D)。我们的发现不排除其他ROS介导的聚集机制的可能性,例如转谷氨酰胺酶3(TGM3)的激活(45-47);我们对微核的蛋白质组学分析在CHMP7结合伴侣中识别了TGM3,但其相互作用不受ROS影响。
为了测试CHMP7的半胱氨酸残基是否参与ROS诱导的微核破裂,我们生成了一个所有CHMP7半胱氨酸(C187、C191、C265、C273和C355)都被丝氨酸残基替代的CHMP7突变体(CHMP7 Cys*)(图3P)。在CHMP7-KO细胞中表达此突变体消除了ROS和NAC对微核破裂的影响(图3,Q和R)。总之,这些观察结果支持了这样一个假设:属于CHMP7的半胱氨酸残基与线粒体衍生的H2O2反应形成二硫键连接的寡聚体,这些寡聚体与LEMD2结合进一步破坏微核膜(图S14F)。这种相互作用似乎是通过一种与生理性CHMP7-LEMD2结合不同的机制发生的(48),此后称为“病理性相互作用”。
微核中的膜修复缺陷
微核与初级核的另一个重要区别是膜在破裂后修复的效率。活细胞成像显示,在跟踪的187个微核中只有一次修复事件,使用了五个独立的微核破裂标记物(图S15A)。尽管ESCRT-III介导的修复在初级核中更有效(1, 49),但Martin等人(50)最近的证据表明,选择性ROS介导的自噬因子和泛素结合蛋白p62(由SQSTM1编码)招募到微核抑制了正常的ESCRT-III功能,阻碍了任何修复的机会。与这一证据一致,H2O2减少了ESCRT-III蛋白的细胞水平,特别是CHMP2A,但这种效应在p62耗尽后得到挽救(图S15,B到D)。类似地,p62缺失增加了CHMP7+微核的数量。然而,病理性ROS介导的CHMP7在微核中的积累与p62无关(图S15,E到G),这表明p62对CHMP7水平的影响可能是通过其ESCRT-III相关功能实现的。因此,癌细胞中的微核可能因ROS依赖的CHMP7-LEMD2结合增加和通过p62介导的经典ESCRT-III功能抑制而减少修复而遭受更多破裂,这可能解释了微核塌陷的不可逆性(图S15H)。
ROS促进了依赖微核的染色体粉碎
微核破裂的一个后果是广泛的DNA损伤,导致封闭的染色体粉碎(14, 51, 52)。这些染色体片段随后以容易出错的方式修复,导致复杂的染色体重排,也称为染色质粉碎(17, 52-54)。为了确定ROS是否作为染色质粉碎中间步骤促进染色体粉碎,我们使用在染色体稳定的DLD-1结直肠癌细胞中建立的多西环素和植物生长素(DOX/IAA)诱导的Y染色体错误分离系统来生成特定染色体的微核(52, 53)。如预期的那样,用ROS处理促进了含有Y染色体的微核破裂,而暴露于NAC则减少了破裂(图S16,A和B)。在DOX/IAA处理后,通过DNA荧光原位杂交分析的有丝分裂展开中多达约20%含有明显粉碎的Y染色体(图S16,C和D)。通过NAC治疗或小干扰RNA介导的CHMP7消耗显著抑制了染色体粉碎(图S16D)。因此,抑制基础ROS或失去CHMP7防止了由于微核破裂导致的染色体粉碎。
缺氧衍生的ROS促进了微核塌陷
肿瘤缺氧是癌症中常见的ROS原因。缺氧通常与氧气可用性减少相关,这被一氧化氮在其结合位点上替代了线粒体ETC复合物IV中的氧气,阻断了电子传输。由此增加的从复合物I和III泄漏的电子负责产生ROS,如前所述(55)。我们询问缺氧是否通过ROS调节微核完整性。实际上,将U2OS和HeLa细胞培养在1% O2中与在大气条件下生长的细胞相比,显著增加了微核破裂(图4A和图S17A)。在缺氧条件下生长的HeLa细胞显示出微核中CHMP7-GFP定位增加(图S17B)。在缺氧条件下生长的细胞中添加NAC治疗显著挽救了微核破裂(图S17C),支持了缺氧响应下增加的微核破裂是由ROS驱动的观点。为了将缺氧机械地联系到由CHMP7介导的微核破裂,我们在缺氧条件下生长的细胞中进行了CHMP7和LEMD2的条件性KO,并观察到在缺氧条件下微核完整性完全恢复,甚至比在常氧条件下生长的细胞中看到的还要低(图4B)。如预期的那样,通过在CHMP7 KO和LEMD2 KO细胞中分别添加PAM* CHMP7和PAM* LEMD2,恢复了缺氧诱导的微核破裂(图4B)。在CHMP7 KO细胞中表达CHMP7 NES*在常氧条件下显著增加了微核破裂,并消除了缺氧的任何额外影响(图4B)。因此,我们展示了缺氧诱导的微核破裂是由CHMP7介导的。
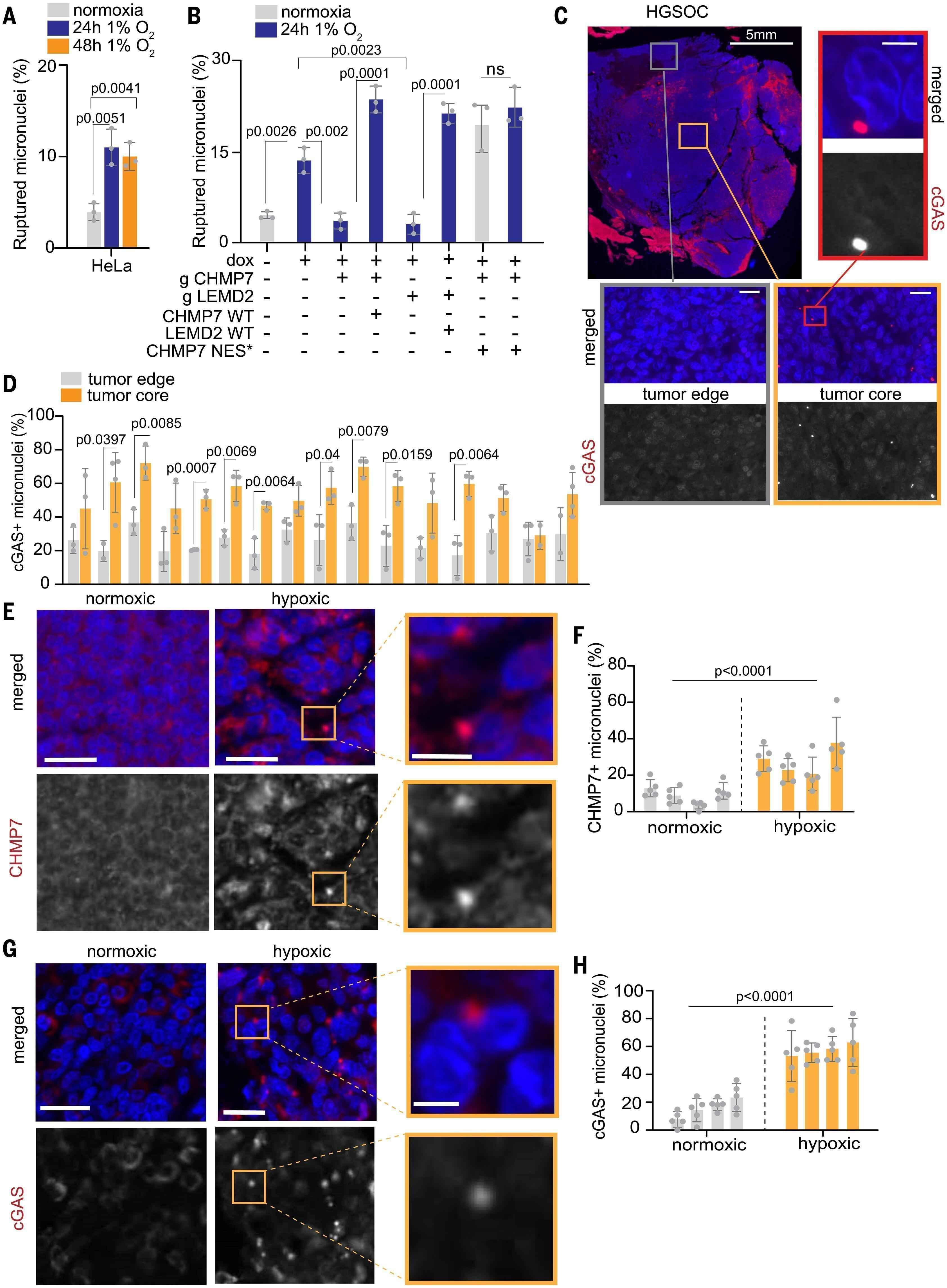
图4. 缺氧衍生的ROS诱导微核塌陷。
(A) 在指定时间的缺氧条件下,HeLa细胞中破裂的MNi。(B) 在大气或1% O2中培养24小时时,用指示蛋白补充和/或耗尽的HeLa iCas9细胞中的破裂MNi。对于(A)和(B),n = 100个MNi,三个生物学重复,平均值±SD,非配对Student's t检验。(C) 高级别浆液性卵巢癌(HGSOC)的代表性图像,用DAPI(蓝色)和cGAS(红色)染色。比例尺,5毫米。(左)放大的肿瘤边缘和肿瘤核心区域部分,假设更缺氧。比例尺,20 μm。(右)来自肿瘤核心的单个微核化细胞的放大图像。比例尺,5 μm。(D) 从16个不同肿瘤的六个感兴趣区域(ROI)中的cGAS+ MNi,如图(E)所示,根据平均ROI距离肿瘤边缘的距离进行划分。距离肿瘤边缘>0.6 mm的ROI标记为“肿瘤核心”,而<0.6 mm的标记为“肿瘤边缘”。非配对Student's t检验,平均值±SD。(E) HPV+头颈癌在常氧或缺氧条件下染色的CHMP7(红色)图像。比例尺,20 μm。(插图)来自缺氧肿瘤的单个微核化细胞。比例尺,7 μm。(F) 每个肿瘤五个ROI中的CHMP7+ MNi,如图(E)所示。通过汇总数据并使用非配对Student's t检验比较常氧与缺氧肿瘤之间的显著性。同样的结果通过单因素方差分析(ANOVA)后跟Tukey多重比较检验得到确认。平均值±SD。(G)HPV+头颈癌染色的cGAS(红色)图像。比例尺,20 μm。(插图)来自缺氧肿瘤的单个微核化细胞。比例尺,7 μm。(H)每个肿瘤五个ROI中的cGAS+ MNi,如图(G)所示。通过汇总数据并使用非配对Student's t检验比较常氧与缺氧肿瘤之间的显著性。通过单因素方差分析(ANOVA)后跟Tukey多重比较检验获得相同的统计结果。平均值±SD。
缺氧与人类癌症中的微核破裂有关
为了检查缺氧对人类肿瘤中微核完整性的影响,我们分析了两个独立的人类高级别浆液性卵巢癌(HGSOC)和人乳头瘤病毒诱导的(HPV+)头颈部鳞状细胞癌(HNSCC)队列。在HGSOC队列中(18),我们研究了微核破裂倾向是否与距离肿瘤核心的远近有关,鉴于增加的缺氧与距离肿瘤边缘的距离有已建立的关系(56)。事实上,我们观察到随着距离肿瘤边缘的增加,微核破裂呈距离依赖性增加,如微核中增加的cGAS染色所证明的(图4,C和D,以及图S17,D和E)。在HNSCC肿瘤队列中,通过18F-氟米索硝唑正电子发射断层扫描成像放射学推断出缺氧,并且将肿瘤分层为常氧或缺氧(57)。不仅缺氧肿瘤显示出更多的CHMP7+微核,而且与常氧对应物相比,它们还表现出三到四倍的微核破裂增加(图4,E和H,以及图S17F),进一步支持了肿瘤缺氧与人类癌症中增加的微核破裂相关的观点。
讨论
我们的数据表明,线粒体与微核的物理接近性使微核暴露于升高的ROS水平。这阻止了CHMP7从微核中输出,同时促进了与LEMD2结合的高级CHMP7结构的形成,破坏了微核膜。这些依赖于ROS的核结构缺陷可能在微核和初级核之间保守存在。然而,大小和曲率的差异、减少的核纤层蛋白水平(16, 20),以及微核中缺陷的核孔复合体(15),可能使后者在较低的ROS阈值下比初级核更容易破裂。
图4. 缺氧衍生的ROS诱导微核塌陷。
(A) 在指定时间的缺氧条件下,HeLa细胞中破裂的MNi。(B) 在大气或1% O2中培养24小时时,用指示蛋白补充和/或耗尽的HeLa iCas9细胞中的破裂MNi。对于(A)和(B),n = 100个MNi,三个生物学重复,平均值±SD,非配对Student's t检验。(C) 高级别浆液性卵巢癌(HGSOC)的代表性图像,用DAPI(蓝色)和cGAS(红色)染色。比例尺,5毫米。(左)放大的肿瘤边缘和肿瘤核心区域部分,假设更缺氧。比例尺,20 μm。(右)来自肿瘤核心的单个微核化细胞的放大图像。比例尺,5 μm。(D) 从16个不同肿瘤的六个感兴趣区域(ROI)中的cGAS+ MNi,如图(E)所示,根据平均ROI距离肿瘤边缘的距离进行划分。距离肿瘤边缘>0.6 mm的ROI标记为“肿瘤核心”,而<0.6 mm的标记为“肿瘤边缘”。非配对Student's t检验,平均值±SD。(E) HPV+头颈癌在常氧或缺氧条件下染色的CHMP7(红色)图像。比例尺,20 μm。(插图)来自缺氧肿瘤的单个微核化细胞。比例尺,7 μm。(F) 每个肿瘤五个ROI中的CHMP7+ MNi,如图(E)所示。通过汇总数据并使用非配对Student's t检验比较常氧与缺氧肿瘤之间的显著性。同样的结果通过单因素方差分析(ANOVA)后跟Tukey多重比较检验得到确认。平均值±SD。(G)HPV+头颈癌染色的cGAS(红色)图像。比例尺,20 μm。(插图)来自缺氧肿瘤的单个微核化细胞。比例尺,7 μm。(H)每个肿瘤五个ROI中的cGAS+ MNi,如图(G)所示。通过汇总数据并使用非配对Student's t检验比较常氧与缺氧肿瘤之间的显著性。通过单因素方差分析(ANOVA)后跟Tukey多重比较检验获得相同的统计结果。平均值±SD。
缺氧与人类癌症中的微核破裂有关
为了检查缺氧对人类肿瘤中微核完整性的影响,我们分析了两个独立的人类高级别浆液性卵巢癌(HGSOC)和人乳头瘤病毒诱导的(HPV+)头颈部鳞状细胞癌(HNSCC)队列。在HGSOC队列中(18),我们研究了微核破裂倾向是否与距离肿瘤核心的远近有关,鉴于增加的缺氧与距离肿瘤边缘的距离有已建立的关系(56)。事实上,我们观察到随着距离肿瘤边缘的增加,微核破裂呈距离依赖性增加,如微核中增加的cGAS染色所证明的(图4,C和D,以及图S17,D和E)。在HNSCC肿瘤队列中,通过18F-氟米索硝唑正电子发射断层扫描成像放射学推断出缺氧,并且将肿瘤分层为常氧或缺氧(57)。不仅缺氧肿瘤显示出更多的CHMP7+微核,而且与常氧对应物相比,它们还表现出三到四倍的微核破裂增加(图4,E和H,以及图S17F),进一步支持了肿瘤缺氧与人类癌症中增加的微核破裂相关的观点。
讨论
我们的数据表明,线粒体与微核的物理接近性使微核暴露于升高的ROS水平。这阻止了CHMP7从微核中输出,同时促进了与LEMD2结合的高级CHMP7结构的形成,破坏了微核膜。这些依赖于ROS的核结构缺陷可能在微核和初级核之间保守存在。然而,大小和曲率的差异、减少的核纤层蛋白水平(16, 20),以及微核中缺陷的核孔复合体(15),可能使后者在较低的ROS阈值下比初级核更容易破裂。
https://blog.sciencenet.cn/blog-41174-1448875.html
上一篇:肠道细菌代谢有机致癌物引发膀胱肿瘤
下一篇:能独立科学研究的AI来了!