博文
Discovery Studio文献解读 | 结核杆菌FtsZ抑制剂计算药理学研究
|

一、文献解读
研究背景:
结核病(Tuberculosis, TB)作为全球致死率最高的感染性疾病之一,其防控面临多重耐药(MDR)与广泛耐药(XDR)菌株蔓延的严峻挑战。世界卫生组织数据显示,TB每年新增约1000万病例,导致150万人死亡,而传统直接督导短程化疗(DOTS)方案(含异烟肼、利福平、吡嗪酰胺、乙胺丁醇)对耐药菌株疗效显著下降,亟需开发作用于新靶点的抗结核药物。近期发表于《Scientific Reports》的研究以结核杆菌细胞分裂蛋白FtsZ为靶点,通过计算药理学方法系统筛选50种新型1,2-二取代苯并咪唑衍生物,明确其作为FtsZ抑制剂的潜力,为抗结核新药研发提供关键理论依据与候选分子。
靶点FtsZ的生物学意义:
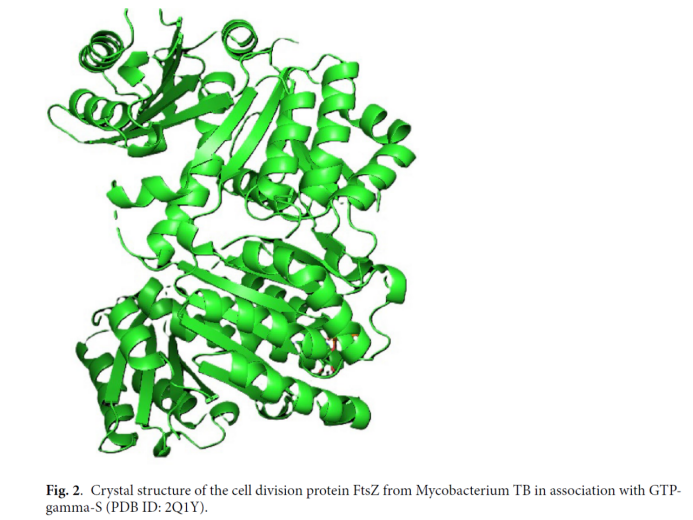
FtsZ蛋白是结核杆菌细胞分裂的核心调控因子,通过组装成“收缩环”驱动细菌缢缩分裂,是细菌增殖的“必需靶点”。与传统抗结核药物靶点(如InhA、KatG)相比,FtsZ不易产生耐药性,且人类细胞中无同源蛋白,具备高选择性优势。本研究选用FtsZ与GTP-γ-S的复合物晶体结构(PDB ID: 2Q1Y)作为对接靶点,该结构清晰呈现活性口袋构象,为分子对接提供可靠三维模型。
苯并咪唑衍生物的药理优势:
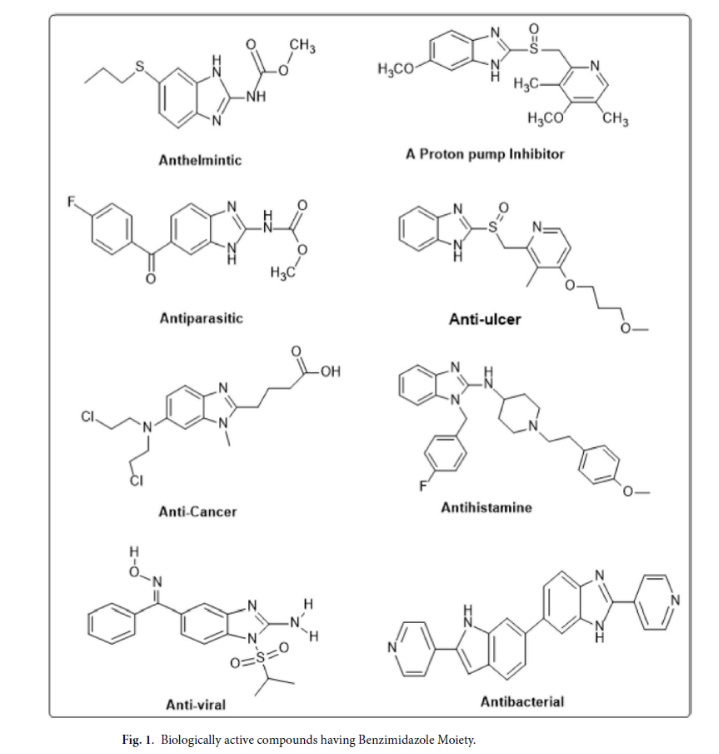
苯并咪唑类化合物是经典的“优势杂环骨架”,具有广谱药理活性,包括抗结核、抗菌、抗病毒等,且其结构易修饰、毒性低,已广泛应用于上市药物。研究表明,1,2-二取代苯并咪唑衍生物可通过特异性结合FtsZ活性口袋抑制其功能,但此前缺乏“大样本筛选+系统ADME评估”的研究,难以明确其成药潜力。本研究正是针对这一空白,构建50种衍生物库进行系统性分析。
Discovery Studio多维度计算药理学技术:
1、靶点预处理
通过BIOVIA Discovery Studio 2021移除FtsZ(PDB ID:2Q1Y)结构中的水分子与天然配体GTP-γ-S,补充氢原子后采用能量最小化(基于MMFF94力场)稳定构象,依据原GTP-γ-S结合位点定义活性口袋。设置对接活性位点中心坐标为x=-6.307、y=53.272、z=0.296,对接半径设为8。以结合能(kcal/mol)为核心指标,结合Discovery Studio可视化分析对接构象,重点关注氢键、疏水作用等关键分子间相互作用。同时,以异烟肼、对氨基水杨酸(PAS)为阳性对照,验证衍生物的结合优势。
2、In silico ADME评估
依据Lipinski五规则(分子质量<500 Da、logP<5、氢键供体≤5、氢键受体≤10)判断成药潜力,通过对比50种衍生物的结构差异(如取代基类型、位置)与对接分数、ADME参数的关联性,明确影响活性与成药性质的关键结构特征,为后续分子优化提供依据。
本文研究结果:高活性候选药与关键规律的发现
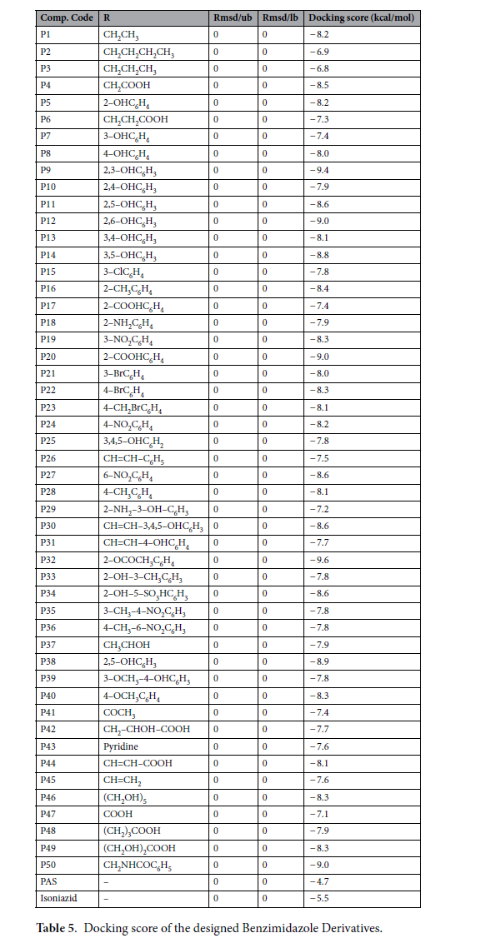
1. 分子对接结果:P32展现最优结合活性
50种1,2-二取代苯并咪唑衍生物的对接分数范围为-6.8~-9.6 kcal/mol,其中12种衍生物(如P9、P12、P20、P32)的结合能低于-8.5 kcal/mol,显著优于阳性对照(异烟肼:-5.5 kcal/mol;PAS:-4.7 kcal/mol)。
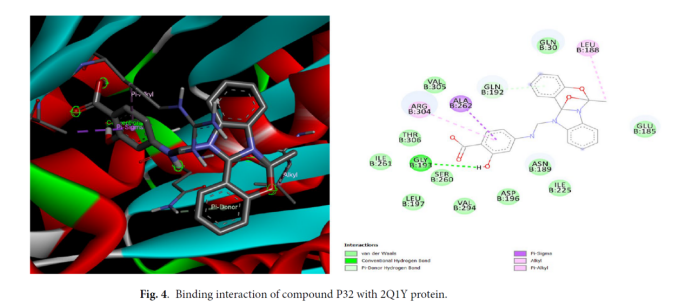
化合物P32(2-OCOCH₃C₆H₄取代)表现最优,对接分数达-9.6 kcal/mol,其结合机制如下:氢键作用:P32的氧原子与FtsZ的Leu188(B)、Gln192(B)氨基形成2条特异性。氢键疏水作用:P32的苯环与Ala262(B)、Gly193(B)的疏水侧链形成稳定疏水口袋。构象适配性:P32分子完全嵌入FtsZ活性口袋,无明显空间位阻,整体构象能量最低。
2. ADME评估结果:多数衍生物具备成药潜力
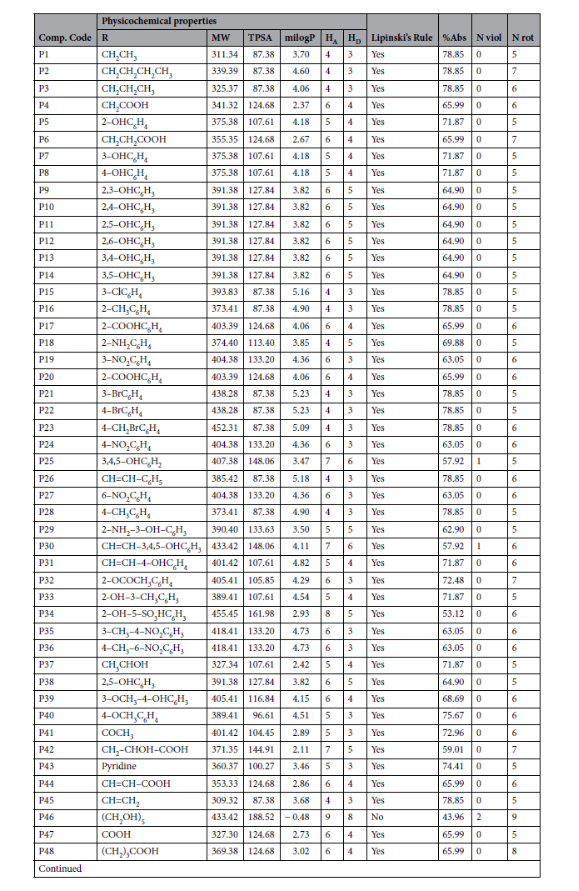
药物相似性:92%的衍生物符合Lipinski五规则(0~1项规则违反),仅P46((CH₂OH)₅取代)因分子质量>500 Da、氢键供体>5违反2项规则,排除为候选药;
吸收性质:胃肠道吸收率普遍高于50%,其中P32吸收率达72.48%,PAS、异烟肼吸收率分别为80.17%、85.54%,说明衍生物口服生物利用度与临床药物相当;
安全性与分布:所有衍生物均不穿透血脑屏障(BBB permeation: No),可避免中枢神经系统副作用;水溶性以“中等可溶性”为主,含羧基(如P4、P6)、羟基(如P5、P7)的衍生物水溶性更佳。
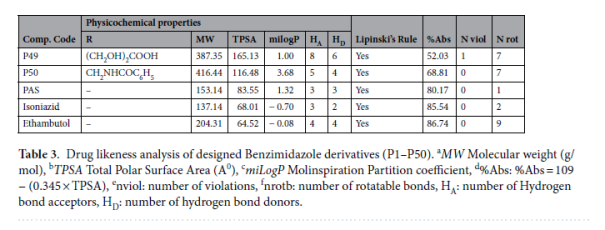
3. SAR分析:明确结构优化方向
通过结构-活性/性质关联分析,总结出3条关键规律: 1,2-二取代苯并咪唑核心为必需骨架:破坏该核心(如移除2位取代基)会导致对接分数下降2~3 kcal/mol,说明核心结构是与FtsZ活性口袋结合的基础; 电负性取代基增强活性:苯环引入Cl、Br等电负性基团可增强疏水作用,提升结合能(如P15、P21对接分数分别为-7.8、-8.0 kcal/mol),但过量会降低水溶性; 乙酰氧基为最优取代基:含-OCOCH₃的衍生物(如P32)在结合活性(-9.6 kcal/mol)、胃肠道吸收(72.48%)、水溶性(中等)间达到最佳平衡,是后续结构优化的首选取代基。
研究与讨论
1、研究的创新点与学术价值
首次建立“大样本1,2-二取代苯并咪唑衍生物+ADME指导的FtsZ抑制剂筛选”范式,相比此前研究,如Dhameliya et al. 2022仅对接无ADME评估、Rode et al. 2020样本量不足,结果更具成药指导意义。 P32等衍生物兼具高FtsZ结合活性与优良药代性质,为抗结核新药研发提供直接候选分子。明确的SAR规律为苯并咪唑类FtsZ抑制剂的结构优化提供“可复制、可推广”的理论依据。
2、研究局限与未来方向
本研究为计算药理学研究,存在一定局限:计算预测结果需通过体外抑菌实验(如MIC测定)验证衍生物对结核杆菌的实际抑制活性,再通过体内动物实验评估安全性与药效。未来可基于SAR规律进一步修饰P32结构(如在苯环引入1个羟基),平衡活性与水溶性,提升成药潜力。
3、结论
本研究通过分子对接、ADME评估与SAR分析,证实1,2-二取代苯并咪唑衍生物是潜在的FtsZ抑制剂,其中化合物P32因最优结合活性(-9.6 kcal/mol)与优良药代性质,成为抗结核新药研发的核心候选分子。研究构建的计算筛选流程与SAR规律,为抗结核药物的高效研发提供了重要参考,也为苯并咪唑类化合物在抗感染领域的应用拓展了新方向。
为什么选择Discovery Studio?
1. Discovery Studio提供一整套同源建模的工具,使用DS,可以轻松完成所有的同源建模过程,包括模板识别、序列比对、自动建模、模型的评估与优化;
2. Discovery Studio中同源建模工具可以预测多种类型生物大分子的结构,包括:球蛋白、抗体、跨膜蛋白等;
3. Discovery Studio中同源建模的核心程序是MODELER,该算法非常经典,目前已发表成百上千的学术文章,学术结果遍布各类杂志,引用率极高;
4. Discovery Studio中分子动力学模拟采用的是经典而强大的CHARMm计算引擎及CHARMm系列力场;
5. Discovery Studio中分子动力学模拟结果可以进行完整便捷的模拟轨迹分析;
6. Discovery Studio中分子动力学模拟可研究对象丰富不单一,如蛋白质、核酸、多糖、多肽、药物小分子以及相应的复合物的模拟;
7. Discovery Studio中分子动力学模拟涵盖领域广泛,如生命、材料等学科。
8. Discovery Studio应用广泛,操作简便,图形化界面十分友好,结果易于分析。
参考文献:Sonwane, P.N., Kumbhare, M.R. Molecular docking and pharmacokinetics of benzimidazole-based FtsZ inhibitors for tuberculosis. Sci Rep 15, 35270 (2025).
二、软件问题咨询
点击下方链接,可对相关软件问题进行咨询。
https://mp.weixin.qq.com/s/y4FHVAGPyoN8q2f-sPcSfA
End

https://blog.sciencenet.cn/blog-3536821-1519540.html
上一篇:Materials Studio | 新能源车续航2000km不是梦 Nat.Commun重磅发布 高能量密度锂硫电池
下一篇:Materials Studio文献解读 | 重庆大学 | Al-5Ti-B细化剂为何在Al-Si合金中失效?
