博文
7年前提出的概念终于被学术界接受了,慨之
||
本周一上午,我看到订阅的新论文提醒邮件有一篇新上线的 Chemical Reviews 论文,综述了单分子力谱在膜蛋白折叠上的进展[1] ——这是我博士论文的课题,虽然毕业之后我就没有继续这个研究方向,仍然习惯性地瞟了一眼论文的配图。
都是眼熟的图片,7年了,还是那几篇经典文献的配图——这个单分子生物物理学的小领域的进展很有限啊。等等,怎么有一张图,格外,诶,我的图?居然从我发在 Biophys J 的论文上截了图,居然还有我的补充材料图,和 Science 论文截图并列一起讨论?Biophys J 影响因子已经掉到3分,几年前就被中国科学院划分到了二区。
原来作者花了一整个 subsection 讨论我和导师 Tobin 在7年前提出的概念 [2]。我们通过计算模拟而提出的概念,终于被学术界的实验课题组接受了,慨之……
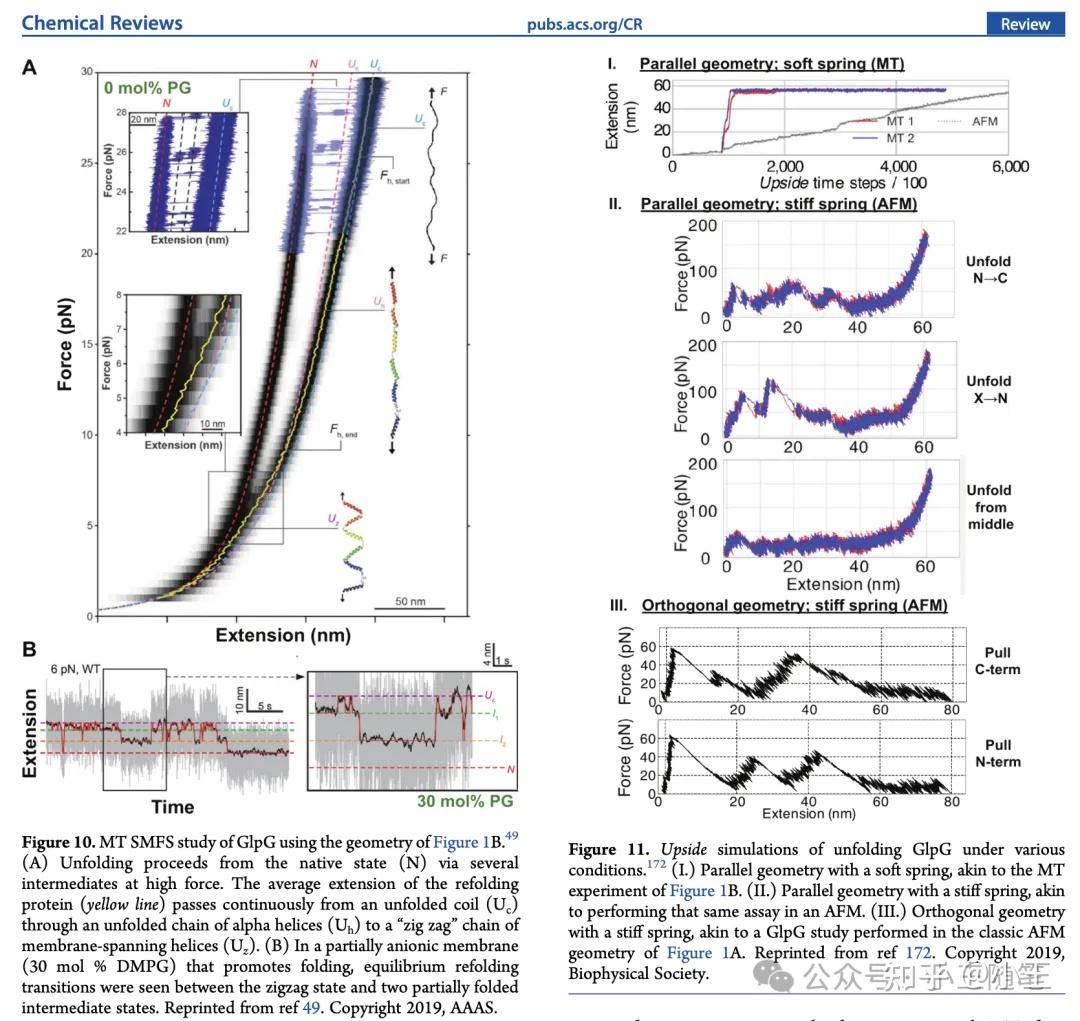
综述[1]截图:右侧图片来自我的论文 Wang et al. Biophys J (2019) [2],左侧图片来自 Choi et al. Science (2019) [3]。
所谓“单分子力谱”(single-molecule force spectroscopy,SMFS),是单分子技术的一种,用原子力显微镜(AFM)、磁镊(magnetic tweezers, MT)、光镊(optical tweezers,OT)等工具,在单分子水平上,操控生物大分子,如蛋白质或RNA。特别地,我们可以用 AFM、MT 等连接在单个蛋白质分子的一端或者两端,而后施加外力,令蛋白质“去折叠”(unfold),而后还可以反向运动,观察蛋白质“重折叠”(refold)。
1998年左右,单分子力谱在刚出现时是一项非常前沿、尖端的技术——是名副其实地字面意思上的“尖端”,因为需要将单个蛋白质分子挂载到 AFM 的探针(tip)尖端,是纳米级、十纳米级的操作。
可以想象,以一定模式(比如匀速)移动 AFM 的探针,对蛋白质施加拉力,会破坏蛋白质的机械稳定性。一方面,探针的形变对应的施加外力被记录下来,另一方面蛋白质分子逐步被拉开,即去折叠,其延展的距离也被记录下来——这样我们就能获得一个 Force-Extension Curve(即力距曲线,即 FEC),这就是“力谱”。
力和延展距离的关系可以用经典的高分子物理学的“蠕虫链状模型”(worm-like chain model)描述——这是单分子力谱的理论基础。
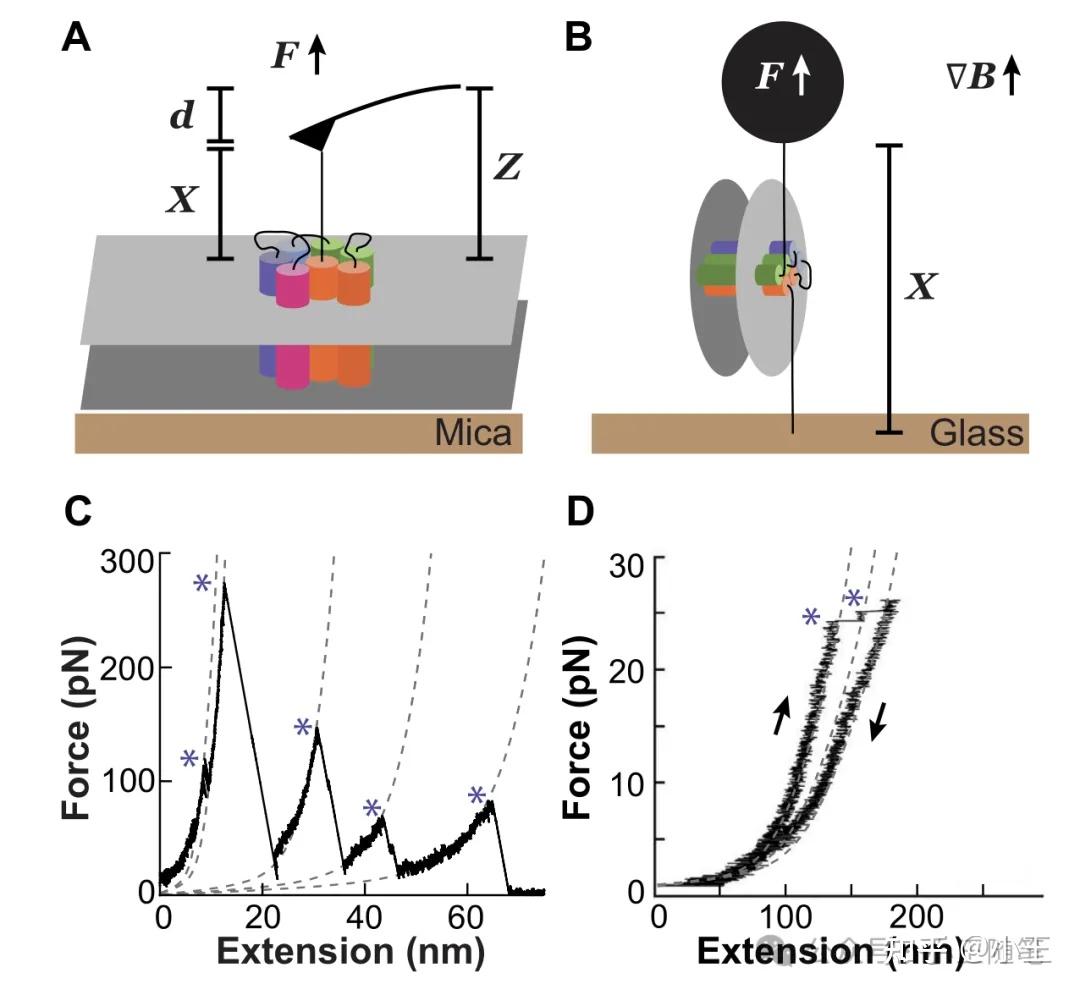
综述[1]图1。(A)AFM 连接跨膜蛋白,纵向牵拉,获得(C) 力距曲线 FEC;(B)磁镊连接跨膜蛋白,横向牵拉(相对于膜平面),获得(D) 力距曲线 FEC。
初始,人们发现单分子力谱这项技术用于研究肌联蛋白(titin)非常合适——肌联蛋白是骨骼肌纤维的一种主要蛋白,其收缩和舒张与机械力息息相关。
很快,2004年,德国的 Daniel J. Müller 课题组基于AFM的单分子力谱研究一种经典7重跨膜蛋白,细菌视紫红质(bacteriorhodopsin,bR),的去折叠和重折叠过程 [4]。这有其独特的时代背景:跨膜蛋白质的插入细胞膜、折叠的过程和机制不清晰,人们转而以“曲线救国”的方式,研究跨膜蛋白的去折叠过程,寄望于能借此逆推折叠过程。
Müller 课题组获得了很有趣的结果,7重跨膜蛋白 bR 的7个跨膜螺旋两两一组被逐步拉开,从嵌入在细胞膜的状态被拉出,成为延展的去折叠链(unfolded chain);每一组2条螺旋被从膜内拉出过程的 FEC (黑色曲线)能非常好地与 WLC 模型拟合(红色曲线)。这个过程中,我们还能看到若干中间态。
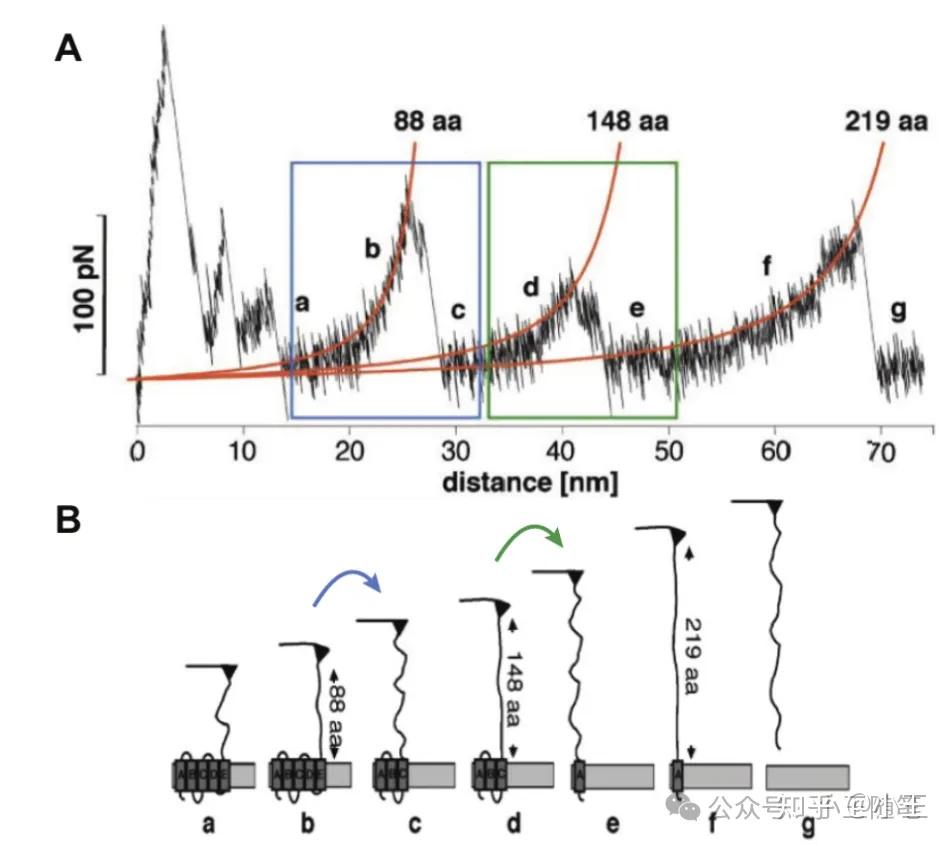 来自论文[4](H. Janovjak et al. Structure, 2004)。
来自论文[4](H. Janovjak et al. Structure, 2004)。
使用 AFM 去折叠 bR 的研究的登峰造极者是科罗拉多大学博尔德分校的 Tom Perkins 课题组。
2017年,Perkins 组的博后余昊(中科大本科04级)发表了一篇 Science 论文 [5],将 AFM 去折叠 bR 的精度推进到了残基尺度,甚至能在拉扯中,看到蛋白质与AFM 的角力——蛋白质抗拒机械力,被去折叠,又反复折叠。下图的一种颜色的FEC 代表一次实验,底部小框放大图内的红色曲线显示出蛋白质的“一圈螺旋”(约3.6个残基)反复去折叠、重折叠。
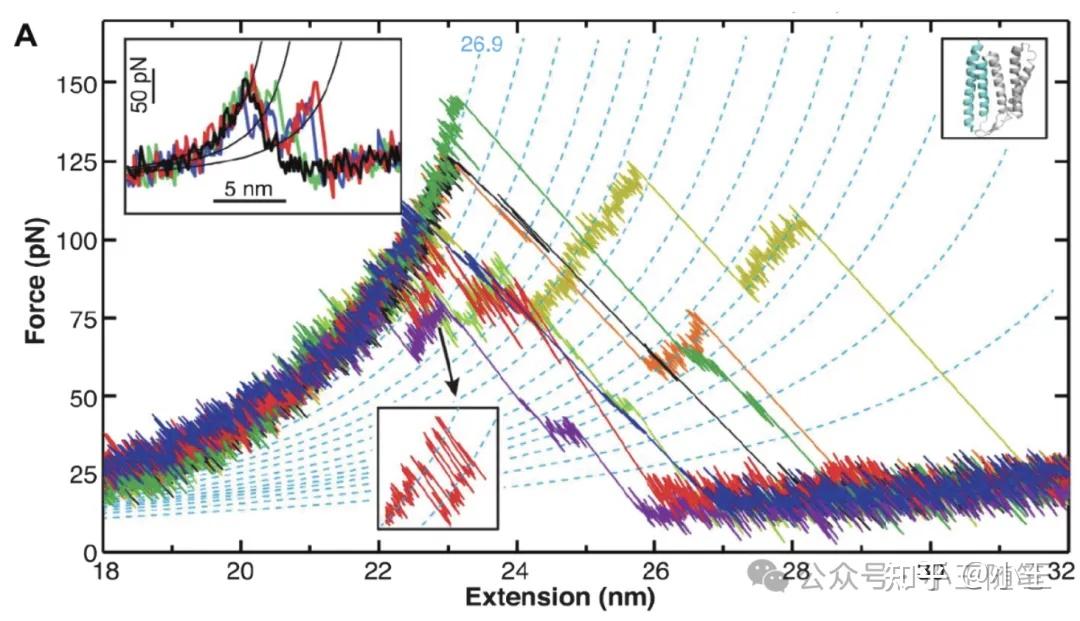
来自论文[5] (Yu et al. Science, 2017)。
这就像是我们用力去拉拽一个弹簧,弹簧不屈服,尽管被外力牵张,但只要施力者稍微松懈,就又回复。精妙的实验!太精妙了!——这是当年处于读博第五年却苦苦无法毕业的我读到这篇论文的第一反应。
与此同时,重复实验显示了不同的去折叠轨迹,意味着 bR 在去折叠过程中经历了多个中间态和多个路径。如果我们希望通过单分子力谱逆向研究蛋白质的折叠,那么直到这篇论文,单分子力谱才真正意义上有了足够精度来触及跨膜蛋白折叠的复杂性。时至今日,我依然认为,这篇 Science 论文是单分子力谱领域近30年(1998至今)的发展史里的巅峰。
我当年的第二反应是,我要模拟它!
其实,模拟 AFM 拉蛋白质,可以说“古已有之”。早在90年代末这项实验技术刚刚诞生时,UIUC的分子动力学巨佬舒克劳(Klaus Schulten)课题组就已经做过了。大家可以想象,它的困难主要在于当年捉襟见肘的算力,因为伴随着拉伸过程,溶剂化水盒子需要同步增大;另一方面,如果拉动速度过快(当时的分子动力学模拟条件下,模拟的拉动速度是实验拉动速度的10的9次方倍),则会超出物理模型的适用范围,力距关系会改变,从而不能可靠地解释实验现象和预测实验结果。
传统的手段不行,我们需要一种超快且高精度的分子动力学模拟方法。
幸运的是,我们有 Jumper。
Jumper 在2017年1月初完成了博士论文答辩,课题是《利用严格机器学习的粗粒化蛋白质折叠和动力学的新方法》。简单地说,Jumper 在读博的5年里独立开发了一种超快且高精度的分子动力学模拟方法,名为 Upside。
我在波士顿举行的“戈登研究会议之膜蛋白折叠”了解到余昊和 Perkins 的工作,当我返回芝加哥决定模拟它时,已经是2017年7月,距离 Jumper 前往 DeepMind 不足两月。
我用一周完成了代码,在 Upside 上添加了一个模块,而后请 Jumper 检查,保证代码风格和 Upside 整体一致且逻辑正确。紧接着,我又用了一周时间获得了最初的结果,与实验[5]看上去“很像”。那一刻,我知道我能毕业了。随后的研究又花去我将近一年半,直到2018年底。

我和导师 Tobin 在7年前提出的概念是什么呢?
我们模拟了两个实验工作,一是2017年余昊和 Perkins 发表在 Science 上的论文,用 AFM 去折叠 bR [5],二是2015年韩国人 Min Duyoung 发表在 Nat Chem Biol 上的论文,用磁镊去折叠 GlpG(6重跨膜螺旋蛋白)[6](Min 后来和卢培龙合作完成了多重扩膜螺旋蛋白的从头设计,Lu, Min et al. Science 2018)。
一方面,我们成功模拟了这两个实验,几乎看到了其所有的中间态;另一方面,我们又看到了更多的,甚至完全不同的去折叠路径和中间态。
我们的解释是,亦即刚刚发表的这篇综述所引述的,牵拉的模式不同会导致观察到的蛋白质去折叠结果不同。
要而言之,AFM 的探针“硬”,弹性系数大(~10 pN/nm),拉动蛋白质去折叠时,随着探针上移,力距积累到足够大时,蛋白质的一部分(可能是一个螺旋,或者多个螺旋)“突然”被拉开,延展的链(extended chain)令探针迅速回复,积累的力消失。探针需要继续移动,重新积聚力量,才能拉开下一部分。这样一来,每一次突然拉开,都为蛋白质抵抗探针拉力提供了缓冲时间,因而能看到更多的中间态。
另一方面,磁镊的弹性系数小(~0.0001 pN/nm),拉动蛋白质去折叠时,在力距积累到相同大小时,磁珠的实际移动距离远远大于AFM的探针(~10万倍)。因此,纵然蛋白质的一部分被拉开会让磁珠略微回复,但是磁力几乎不变,依然有一个积聚的较大的力施加在蛋白质上。这种情况下,我们只能看到很少的中间态。
以上两种情况是湿实验中看到的,我们的模拟都能够很好地解释。
而后,我们用完全一致的参数条件,模拟预测了一种湿实验暂时无法做到因而没有观察过的拉动模式,即假设我们能用如 AFM 一样弹性系数大的磁镊来拉动 GlpG 呢?
我们看到了极多的去折叠路径和中间态,GlpG 的去折叠既能从 C 端开始,也能从 N 端开始,还能从中间开始。这意味着,过往的单分子力谱实验所观测到的蛋白质去折叠过程其实是人为观察的结果,不同的实验或观察方式引起不同的坍塌,并不是蛋白质的内禀性质。换句话说,我们认为前面的湿实验的结论,注意是结论而不是技术本身,对于解读蛋白质折叠,没用。
我们的这个结论有点“上车焊门”的味道,不是吗?
因为此,我在毕业后失去了对这个领域的兴趣。并非这项技术无用,例如我们可以利用单分子力谱将延展开的多肽链或者有机分子拉过纳米孔获得指纹谱;而是,我认为通过单分子力谱研究蛋白质的折叠或者去折叠不是一条通途。
我们获得这个预测之初也是困惑而疑虑的,为此,我们和 Biophys J 杂志编辑——正是 Tom Perkins 本人,反复讨论。Perkins 事实上充当了我们论文的第四审稿人,在审稿中提出了最多和最严苛的问题。一些由在校教授担任编辑的老牌杂志其实有着更加严格的审稿要求。
我们强调模拟的参数设置保持一致,是因为我们要做的是 simulation,而不是 modeling。我写过,我的老爷爷导师 Karl 曾经问我二者的区别(见《AlphaFold首席科学家John Jumper荣获2024年诺贝尔化学奖》)。区别是,simulation 模拟不加干涉,设定好初始条件后,任其发展,可以用于预测尚未出现的结果;modeling 模拟添加约束,对照目的地前进,随时调控,可以用于解释已经存在的现象。 计算之于实验的意义正在于:解释 & 预测。
未竟:
已在华中科技大学独立的余昊课题组,在2024年发表了一篇 JACS Au 论文[7],用 AFM 研究二酰甘油激酶(diacylglycerol kinase)的去折叠,并用我们的工具自行做了模拟。他们的结论是:The remarkable agreement between experiments and simulations …… (见下图)。
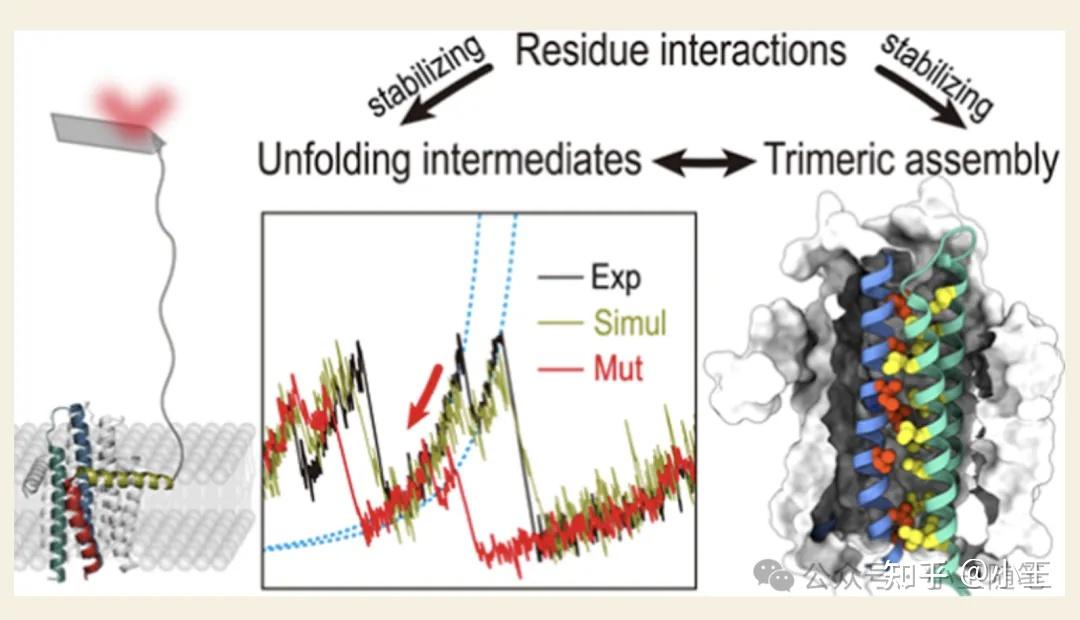
来自论文[7] (Yang et al. JACS Au 2024)。
本文完。
2026.2.11于深圳
参考文献[1] David R. Jacobson. Single Molecule Force Spectroscopy to Probe Intermediates and Energetics of Membrane Protein Folding. Chem Rev (2026). DOI: 10.1021/acs.chemrev.5c00612
[2] Z. Wang, J. M. Jumper, K. F. Freed, T. R. Sosnick. On the Interpretation of Force-Induced Unfolding Studies of Membrane Proteins Using Fast Simulations. Biophys. J. 2019,117, 1429−1441.
[3] Choi et al. Watching helical membrane proteins fold reveals a common N-to-C-terminal folding pathway. Science 2019, 366,1150−1156.
[4] Janovjak et al. Probing the energy landscape of the membrane protein bacteriorhodopsin. Structure 2004,12, 871−879.
[5] Yu et al. Hidden dynamics in the unfolding of individual bacteriorhodopsin proteins. Science 2017, 355,945−950.
[6] Min et al. Mapping the energy landscape for second-stage folding of a single membrane protein. Nat Chem Biol 2015, 11(12):981-7.
[7] Yang et al. Mechanistic Insight into the Mechanical Unfolding of the Integral Membrane Diacylglycerol Kinase. JACS Au 2024, 4, 1422-1435.
https://blog.sciencenet.cn/blog-3458695-1522053.html
上一篇:AlphaFold-Cluster的五回合论战
下一篇:速递:折叠迪斯科Folddisco
