博文
AlphaFold-Cluster的五回合论战
||
AF-Cluster发表于2023.11的Nature,以蛋白质计算领域的发展速度来衡量,已经是老文章了。自其发表以来,正反两方科学家进行了多轮攻辩,过程涉及2篇Nature、1篇Nat Comms、1篇J Mol Biol,以及6篇预印本,互相“中门对狙”,并在2025.8暂告一段落,可谓精彩。更重要的是,这一系列工作所讨论的问题——AlphaFold2/3 预测具有多构象的蛋白质的结构,是AlphaFold的重要议题之一。
下面,我们简单地回顾这场多回合论战:
第一回合:正方,AF-Cluster
第二回合:反方,单序列就够了
第三回合:反方,AlphaFold2是大记忆恢复术
第四回合:反方,随机采样更好
第五回合:正方,你全错
正方:
Hannah K. Wayment-Steele (Scripps & HHMI, UW Madison), Sergey Ovchinnikov (MIT), Lucy Colwell (Google & Cambridge), Dorothee Kern (Scripps & HHMI)
P1. Wayment-Steele, 2023.11, Nature, Predicting multiple conformations via sequence clustering and AlphaFold2.
P2-1. Wayment-Steele, 2024.7, bioRxiv, A resource for comparing AF-Cluster and other AlphaFold2 sampling methods.
P2-2. Wayment-Steele, 2025.3, bioRxiv, Sequence clustering enhances AlphaFold2.
P2-3. Wayment-Steele, 2025.7, bioRxiv, Does Sequence Clustering Confound AlphaFold2?
P2-4. Wayment-Steele, 2025.8, JMB, Does Sequence Clustering Confound AlphaFold2?
其中P1就是AF-Cluster论文,P2-1, 2, 3是P2-4的三个版本的预印本。
正方相关文献:
P3. Wayment-Steele,2024.10, PNAS, The conformational landscape of fold-switcher KaiB is tuned to the circadian rhythm timescale.
谢尔盖(Sergey Ovchinnikov)是诺奖得主 David Baker的博士学生,2017年博士毕业,若非因为他,我大概率不会特别关注这场辩论并细读这些论文。
反方:
Lauren L. Porter课题组(NIH),包括Devlina Chakaravarty、Joseph W. Schafer等。
N1. Chakaravarty, 2023.11, bioRxiv, ColabFold predicts alternative protein structures from single sequences, coevolution unnecessary for AF-cluster.
N2-1. Chakaravarty, 2023.12, bioRxiv, AlphaFold2 has more to learn about protein energy landscape.
N2-2. Chakaravarty, 2024.8, Nat Comms, AlphaFold predictions of fold-switched conformations are driven by structure memorization.
N3-1. Schafer, 2024.1, bioRxiv, Sequence clustering confounds AlphaFold2.
N3-2. Schafer, 2025.2, Nature, Sequence clustering confounds AlphaFold2.
其中N2-1、N3-1分别是N2-2、N3-2的预印本。
反方相关文献:
N4. Schafer, 2023.9, Nat Comms, Evolutionary selection of proteins with two folds.
为了方便起见,本文仅用上述所有文章所提及的KaiB蛋白作为案例来讨论,而不涉及论文中的其它蛋白,除非必要。
第一回合:正方,AF-ClusterWayment-Steele等人在2023.11.13发表了AF-Cluster。
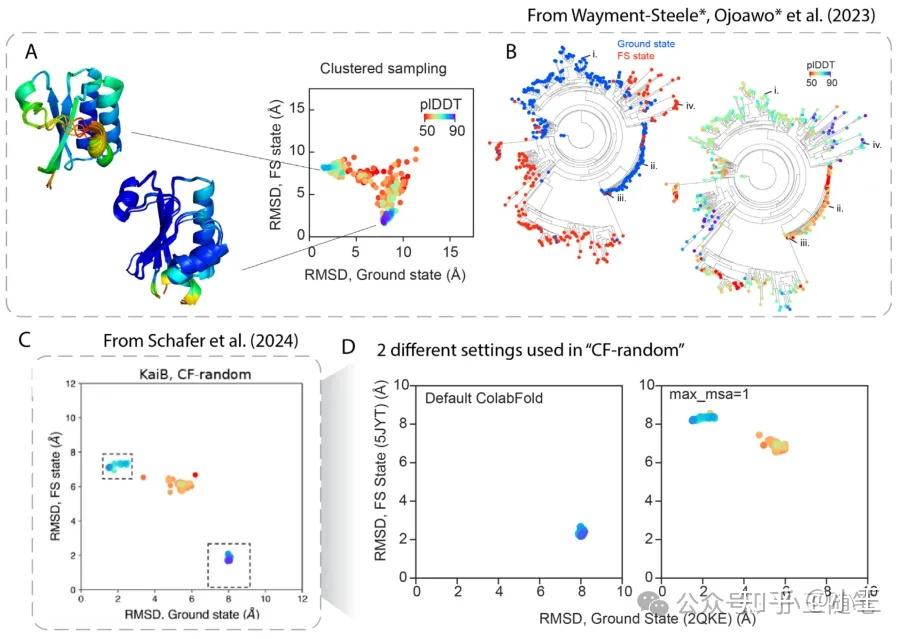
作者研究变构蛋白(metamorphic protein)KaiB家族的预测结构的进化分布。KaiB家族的蛋白质存在两种稳定的实验晶体结构——基态(ground state)和折叠转换态(fold-switched state, FS)。KaiB蛋白仅有108个残基,其C端部分会发生涉及约40个残基二级结构的构象变化,从而在经典的硫氧还蛋白样结构与独特的替代构象之间切换(P1论文图1a,b)。
作者观察到,通过序列相似性而对KaiB家族的蛋白质序列进行聚类,能使AlphaFold2以高置信度对已知变构蛋白的多种状态进行采样(P1论文图1d)。例如,使用全部KaiB家族的蛋白质序列,搜索并构建MSA后,用ColabFold默认设置进行预测,获得FS态(P1论文图1c左);然而,如果在序列聚类中,挑选与已知基态结构的序列最接近的序列来搜索并构建MSA,而后预测,则获得基态(P1论文图1c右)。
因此,作者假设:如果一个蛋白家族在进化中形成了偏好不同构象的亚群,那么这些亚群的序列中应包含指向特定结构的“进化耦合(evolutionary coupling. EC)信号”;所以,不必添加任何先验知识,只要能解耦局部序列的EC信号,就能引导AF2预测出不同的构象。
作者将这种简单的MSA采样方法名为AF-Cluster。在AF-Cluster产生的结构分布中,置信度最高的区域恰好对应实验已知的两种状态(P1论文图1e,f)。
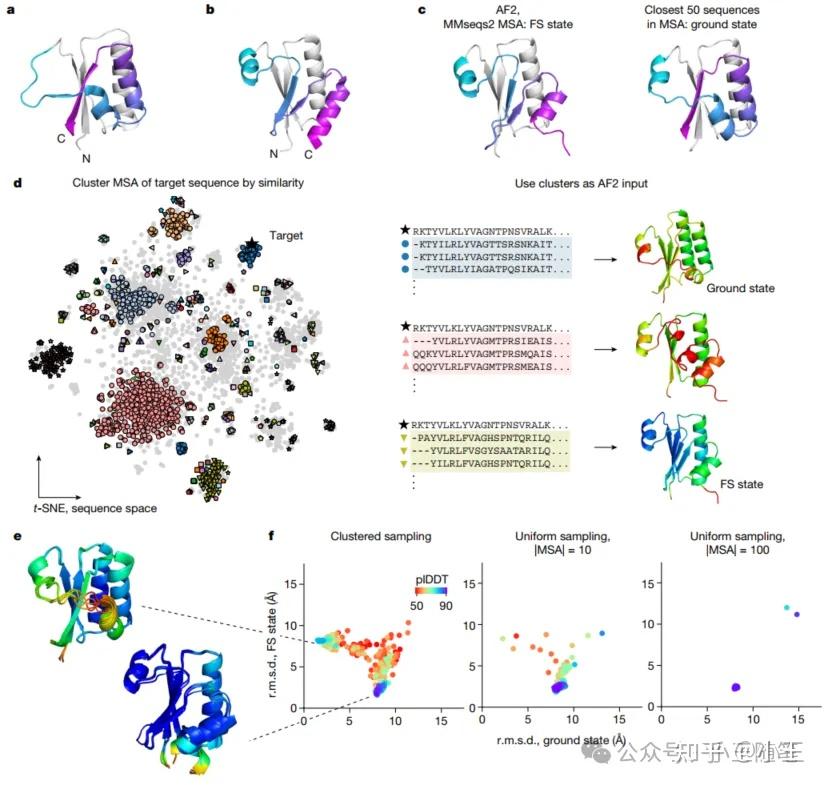
P1论文图1。
真正的亮点在于实验验证:作者预测了一个未经充分研究的KaiB变体(KaiBTV-4)应稳定在FS态,并通过NMR实验证实了这一预测(P1论文图2d,f)。更令人印象深刻的是,作者通过比较偏好不同状态的序列聚类,设计出仅三个突变就成功将KaiBRS从基态翻转至FS态(KaiBRS-3m),实验证实突变体群体的FS构象比例从野生型的11%大幅提升至86%(P1论文图3)。
这里要明确:AF-Cluster指代整个pipeline——给定序列,使用ColabFold内置的MMseqs2搜索UniRef30生成该序列的MSA,去除含超过25%空位的序列,再采用DBSCAN算法,对MSA内的序列进行聚类,而后对每个聚类,仅使用该聚类所含的序列(即原MSA的一个子集)用AF2预测结构;如此,获得一个结构系综。
第二回合:反方,单序列就够了在P1见刊仅仅1周后,Porter团队在2023.11.21预印了N1,直白地说,AF-Cluster错了——进化耦合不是必需的,将单序列喂给ColabFold一样能预测出同样的结果(N1论文图1)。
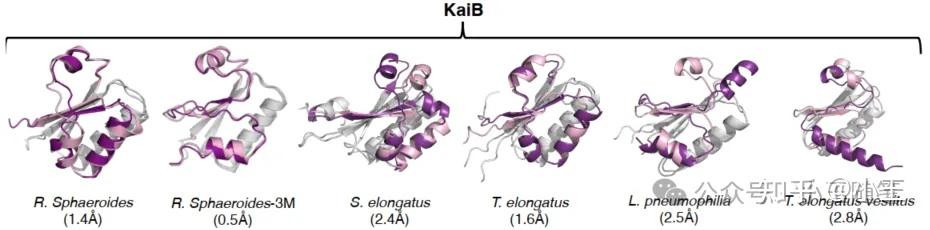
N1论文图1。C端变构区域的残基,使用AF-Cluster预测的着粉色,使用单序列预测的着紫色。
这篇预印非常短(因为只隔了一周嘛),不是一篇完整的研究论文(因此至今停留在预印状态)。
作者使用AF-Cluster预测KaiB蛋白结构,对比仅仅使用单序列(不搜索构建MSA,当然也没有AF-Cluster式的MSA分组采样)的预测结构,发现二者几乎一样,并在观察MSA Transformer产生的接触图后认为不存在进化耦合信号。
第三回合:反方,AlphaFold2是大记忆恢复术仅仅3周后,Porter团队在2023.12.12预印了N2-1,并最终在2024.8.24见刊于Nat Comms,即N2-2。
如果我们是Porter团队,由N1所展示的初步结果,很自然地会引申:既然使用单序列能够预测出和AF-Cluster(使用不同深度的分组后的MSA)一样的结果,那么,针对构象转换(fold-switched)蛋白,AF本质上是否只是一个记忆模型?或者等同地问,AF有没有学习到蛋白质物理(protein energetics)?
N2-2就是为了回答这个问题。
如果AF真正掌握了蛋白质物理,它就应该能够持续且精确地预测折叠转换蛋白的所有实验观测构象;若其无法做到这一点,则意味着某些基于AF的预测很可能主要依赖模式识别——记忆。
为此,Porter团队扩大考察对象的范围,收集了92个经过实验验证的折叠转换蛋白——这是当时最全的经过实验验证的折叠转换蛋白数据集,对AF-Cluster、AlphaFold2/3及其他采样方法进行了大规模基准测试,颇令读者有“宜将剩勇追穷寇”之感。我相信这类“一网打尽”式的基准测试也颇能获得杂志编辑的好感。
Porter团队测试了多个AF模型,包括AF 2.2.0,AF 2.3.1,AFM,AF3等,以及3种“增强MSA采样”的方法——SPEACH_AF、AF-Cluster、ACE。ACE是Porter团队在N4中开发的,全名为“alternative contact enhancement”。结果显示:ALL_AF2的成功率仅为35%(32/92),而AF-Cluster更差——只预测对了18/92组折叠转换蛋白的结构(N2-2论文图1a)。
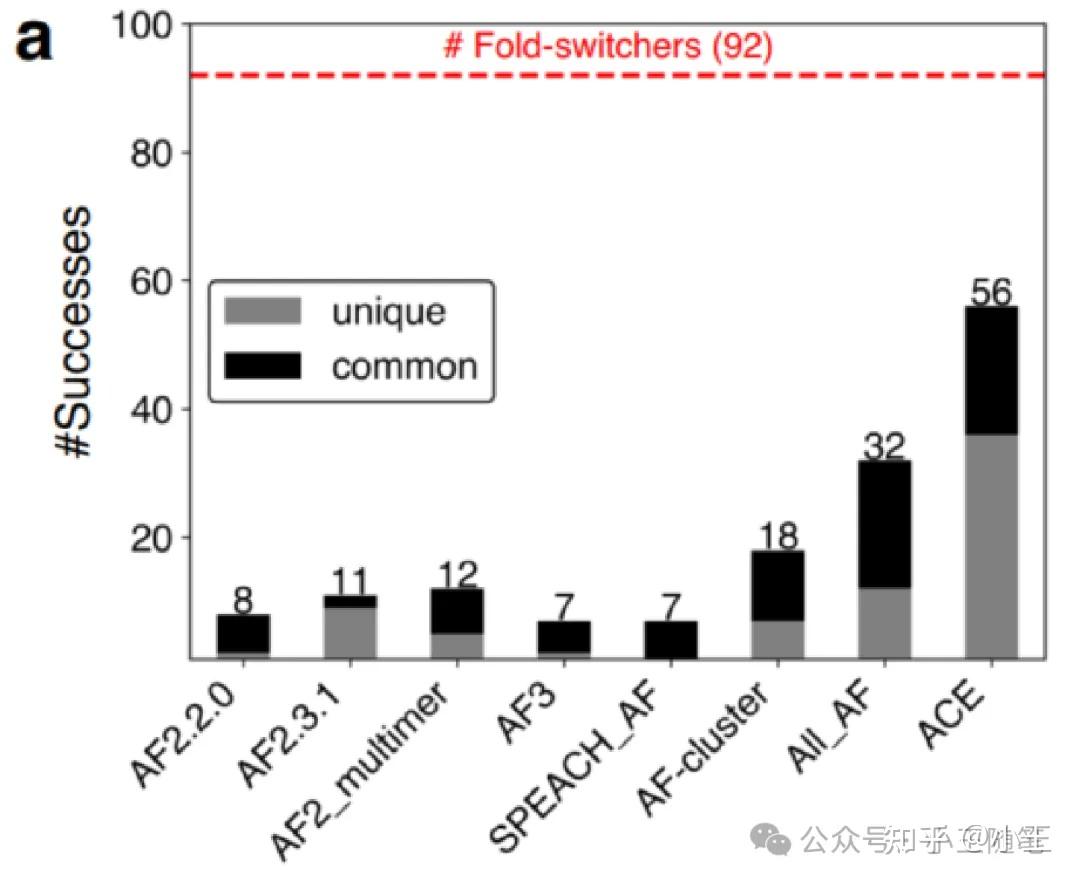
N2-2论文图1a。图中ALL_AF根据N2-2的图片描述,应当为ALL_AF2,指结合了所有AF2模型的结果。
AF-Cluster的表现劣于ALL_AF2让Porter团队认为P1论文的核心结论是错的。否则,如果AF-Cluster真地通过序列聚类而揭示出编码在进化信息中的构象偏好,那么AF-Cluster应当始终优于AF2。在多样化的基准测试中,仅约20%的成功率只能证明AF-Cluster在P1论文中的成功是一种特定场景的偶然现象,而非可推广的普适原理。
第二,Porter团队展示了两个例子,证明AF2.3.1,AF3,AF-Cluster不能正确预测其训练集之外的折叠转换蛋白的结构。这两个例子分别是Sa1和BCCIP。Sa1是一个人工设计蛋白,存在α/β- plait和3αhelix两种实验结构;BCCIPα和BCCIPβ是两种人蛋白质异形体(isoform),二者序列一致性为80%。AF2.3.1,AF3,AF-Cluster不能预测出Sa1的3αhelix构象,及BCCIPα构象。

N2-2论文图3。
考虑到测试集所包含的92组蛋白都在AF2训练集的截止日期之前被上传到PDB,因此很可能被包含在AF2的训练集之中,作者认为,AF的预测依赖记忆。
第三,Porter团队通过直接控制Evoformer的输入而检查AF2的输出,分3步:(1)将MSA输入AF2,但不进行recycling,预测出构象1,这种情况下,AF2的预测结构仅由Evoformer控制,而Evoformer是对进化耦合信息的解读;(2)将MSA输入AF2,进行recycling(即完整AF2管线),预测出构象2;(3)将MSA输入AF2,进行recycling,并在每一轮recycling之后提取Structure Module输出的MSA,将之作为输入饲喂给没有recycling的AF2(如同1),这种情况下每次都预测出构象1。
这个结论非常有意思,特别是(3)!因为(2)的结论,我们知道最后一次循环后Structure Module输出的MSA会被推理为构象2;那么,我们很自然地会认为,每一次循环后,Structure Module输出的MSA会逐渐接近最后一轮的MSA,进而用每一轮循环后的MSA进行推理应该会获得一个由构象1向构象2转换的轨迹 —— AF2论文展示了类似的轨迹。
但是,(3)却显示,至少对Porter团队所考察的折叠转换蛋白(RfaH)而言,这条轨迹其实是一个阶跃函数,直至最后一轮循环,前面所有的取值都是构象1,然后猛地跃迁到构象2。
并且,(1)-(3)综合表明,对折叠转换蛋白RfaH而言,Evoformer从MSA中解读的进化耦合信号唯一地支持构象1,但是完整的AF2管线却强制性地推理出构象2,所以在AF2预测结构中起决定性作用的不是进化耦合信号,而是从训练集中记忆到的模板结构(通过结构模块而体现)。
第四,Porter团队再次利用单序列(而非MSA)进行预测来证明AF2是记忆模型。已知折叠转换蛋白RfaH的实验结构中的C端结构域(CTD)呈现螺旋束,而CTD序列片段的实验结构呈现β折叠。作者只用RfaH序列的CTD部分作为单序列输入,不搜索MSA,也不进行recycling,AF2唯一地预测出螺旋束。作者认为,出现这种结果的原因是AF2在训练中学习、记忆到了由完整RfaH结构所提供的CTD结构模板,而非蛋白质物理。
第四回合:反方,随机采样更好又过了三周,Porter团队在2024.1.5预印了N3-1——剩勇追穷,恐怖如斯,并最终在2025.2.19见刊于Nature,即N3-2。
我们需要指出,N3-2是一篇“matters arising”(对已发表文章的评论或澄清)——应对发表在Nature的P1,不是一篇“research article”,故其篇幅也非常短(其实N3-2可以看做N1的正式见刊版)。
尽管如此,Porter团队在短时间内(从P1见刊到N3-1预印仅7周)提出了一个新方法:CF-Random,使用ColabFold(CF) + 随机采样的浅层MSA预测结构(N3-2论文拓展图2a)。
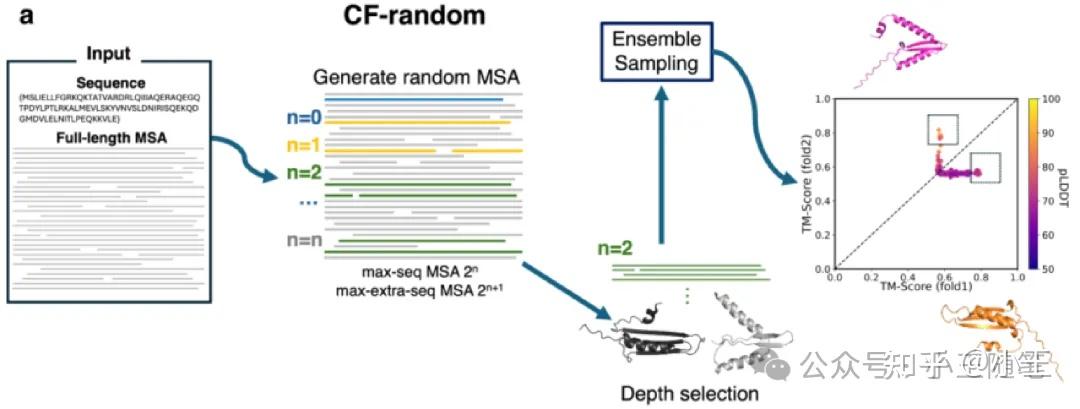
N3-2论文拓展图2a。
N3-2的核心论点是P1(AF-Cluster)不是如N2-2所发现的使用范围有限(仅在约20%的折叠转换蛋白上成功),而是在根本上错误的,即“AF-Cluster通过分离出MSA的局部进化耦合信号来预测折叠转换”是错的——简直是要逼着P1撤稿,是可忍孰不可忍?
Porter团队给出的论点与其在N1中给出的论点是类似的:如果AF-Cluster真地有效,那么它应该比使用随机采样的浅层MSA(N3-2)/ 单序列(N1)预测结果好。但是,结果显示并非如此:CF-Random的表现稳定地好于AF-Cluster(N3-2论文图1b),并且AF-Cluster所考察的MSA中并没有足以预测稳定出不同折叠状态的独特的进化耦合信号(N3-2论文图2a)。
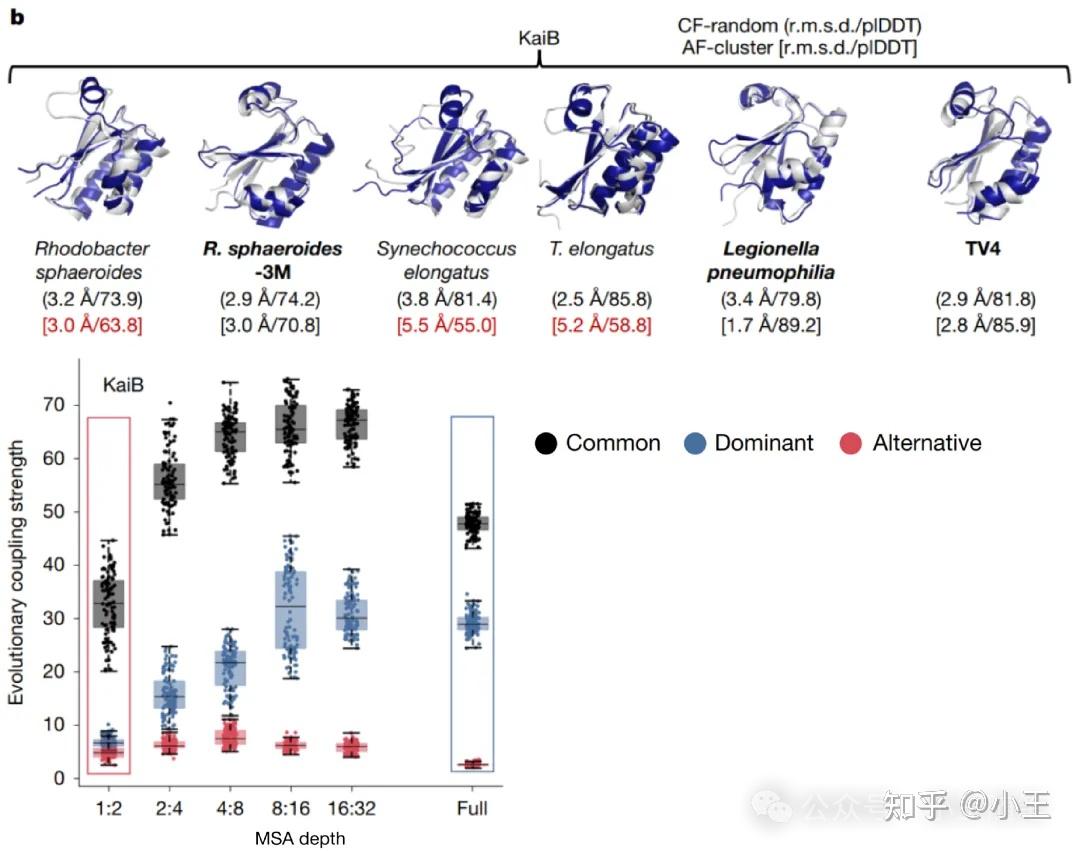
N3-2论文图1b(上),图2a(下)。
图2a展示了CF-random测试的不同MSA深度下,主导构象(dominant)与替代构象(alternative)各自独有或两者共有的(common)进化耦合概率之和。在所有MSA深度中,替代构象独有的进化耦合概率均微乎其微,表明这些耦合并非驱动替代结构预测的主要因素。蓝色与红色方框分别表示用于预测主导构象和替代构象时所采用的MSA深度范围。每个箱线图包含600个数据点,箱体表示四分位距,中线代表中位数,须线延伸至四分位距的1.5倍范围。横坐标轴的X:Y表示法对应输入ColabFold的--max-seq与--max-extra-seq参数值。
第五回合:正方,你全错终于轮到正方为自己辩护,Wayment-Steele等人在2024.7、2025.3、2025.7接连预印P2-1、2、3,多次更新预印本论文,并最终见刊于JMB,即P2-4。
Porter团队的论文取标题《Sequence clustering confounds AlphaFold2》,Wayment-Steele等人回击论文取标题《Does Sequence Clustering Confound AlphaFold2?》,可谓“中门对狙”,文中Wayment-Steele等人逐一反驳了N1、N2-2、N3-2。
第一,针对N1,Porter团队选择性地呈现数据。N1篇幅很短,结论是一句话:使用单序列,无需搜索MSA,更无需AF-Cluster聚类MSA,就能预测出和AF-Cluster等同的结果。
这个结果其实已经包含在P1论文的补充材料中,即Wayment-Steele等人做了质控,证明在大多数情况下使用单序列或者浅层MSA就足够预测出相应的构象(P1补充材料图1d,即P2-4图1A)。——难绷,虚空打靶……
我们姑且将系统发育树上最接近的10条序列构建的MSA称为“浅层MSA”(shallow MSA),这可以认为是MSA聚类之后每个聚类内最接近的10条序列(如果聚类所含序列不少于10条)。
N1认为,对KaiBTV-4,使用单序列或2条序列(构建深度极浅的MSA)就可以获得匹敌使用浅层MSA做预测的结果。
Wayment-Steele等人在P2-4图1B - E展示这是错误的:(1)使用浅层MSA,所有AF2的5个模型均在1次循环内快速收敛到正确的NMR结构,且置信度很高(P2-4图1B,D上排);(2)使用单序列,AF2的5个模型中,有4个即使经过多次循环也不能正确预测,仅1个模型(模型5)在7次循环后接近正确结构,但构象不完整且置信度很低(P2-4图1C,D下排)。——那么,如果没有先验知识,从单序列预测的5个模型中,研究者很可能会选择置信度最高但结构错误的模型3(P2-4图1D下排黑色框)。并且,N1论文中实际挑选出用于展示的所谓正确结构是模型4给出的结果(P2-4图1E),是错误的。
因此,N1的结论全错。

P2-4图1。
第二,针对N2-2,Porter团队的基准测试集的选择不恰当。Wayment-Steele等人在P1中强调,AF-Cluster的假设是有条件的:若一个蛋白质家族的进化历程导致了偏好不同状态(构象)的亚群,则聚类分析可揭示这种构象偏好,即MSA clustering reveals evolutionary encoded structural preferences。在不符合此条件的蛋白质上测试该假设,不是有效的验证。
例如,N2-2中,Porter团队展示了2个反例(我们在上面提到过):人工设计蛋白Sa1,存在α/β- plait和3α helix两种实验结构的BCCIP。AF-Cluster不能正确预测这两例的折叠转换是可预期的。人工设计的Sa1没有自然演化史,你能期待看到进化耦合信号吗?对BCCIP而言,BCCIPα和BCCIPβ的序列,因为可变剪切(alternative splicing)而存在20%完全不同的片段,由BCCIPα序列搜索并构建MSA,这个MSA在BCCIPα序列与BCCIPβ序列不同的区域完全没有覆盖(P2-4图A),这显然也不适用于AF-Cluster的假设。
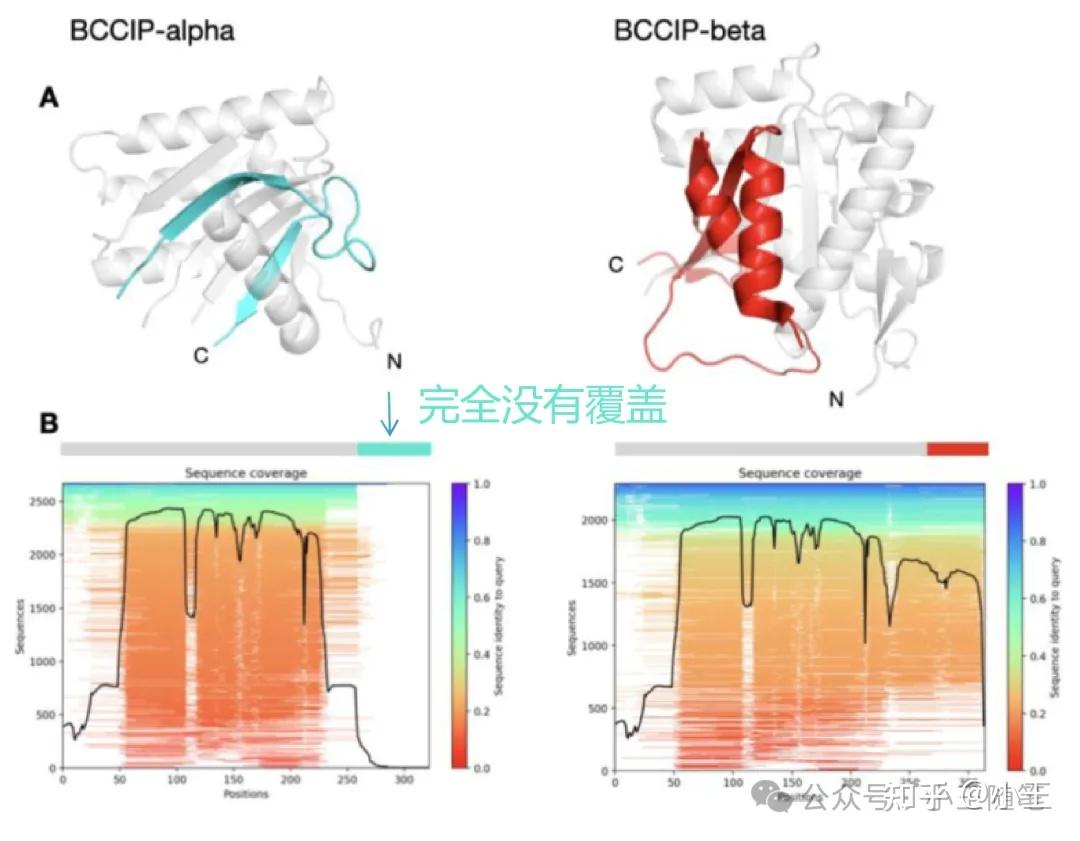
P2-4图A。
那么,我们可以问:N2-2所使用的包含92组折叠转换蛋白的基准测试集有多少组不符合AF-Cluster的适用范围呢?换句话说,虽然同为“折叠转换”,但具体是因为进化中累计的点突变而导致的,还是因为其它方式?
一个方法在自身理论不适用的地方失败,不能用来证明它在适用领域内的机制是错的。All models are wrong, except some are useful. Porter团队的基准测试没有证明AF-Cluster的假设错误,而是为其划定了适用范围。
从这个角度理解,针对N2-2和N3-2,Porter团队误解了AF-Cluster的研究目标。
Porter团队认为AF-Cluster是一种表现很差的变构蛋白的结构预测器,原因是将AF-Cluster默认为一个通用构象预测器,但是AF-Cluster却不够普适、不够稳健。他们的批判逻辑是:“如果一个方法声称能预测构象,它就必须在所有或大多数已知的构象切换蛋白上有效,且其置信度指标必须可靠,且不能产生假阳性。” 因此,他们设计了大规模的基准测试,统计成功率,并检查其特异性。
然而,P1的研究目标是,找到了一个探索蛋白质进化-构象关系的新角度,将AF-Cluster视为一个假设生成与机制探索工具,而后通过严格的湿实验证明它在某些情况下是真实存在的机制。其核心目标是回答:“在这个蛋白家族的进化历程中,是否存在偏好不同构象的序列亚群?如果存在,这些亚群是否编码了特异的构象信号?” 它的成功标准是:在适用的体系(即自然进化产生的构象切换蛋白)中,能否揭示这种信号,并做出可被实验验证的预测。它提供的不是一个简单的“是/否”答案,而是一个序列-结构偏好性的分布(P1图2a、b、c)。
所以,在这个意义下,反方依然在虚空打靶。

P1图2a、b、c。
第三,针对N2-2、N3-2的技术细节,Porter团队没有采用与AF-Cluster(P1)相同的参数设置。简言之,AF-Cluster使用了更新的参数集(model_1_ptm, model_2_ptm 等),而N2-2、N3-2则混用了更老的参数集(model_1, model_2 等),二者产生的结果有很大差异(P2-4图B2)。因此,Porter团队的对照组运行结果(旧参数)和Wayment-Steele等人的AF-Cluster运行结果(新参数)的pLDDT分数不能有效比较,而pLDDT是N2-2基准测试中衡量“置信度”和“成功”的关键指标。——说实话,这挺难绷的,N2-1求速发表,却没有检查好参数设置。
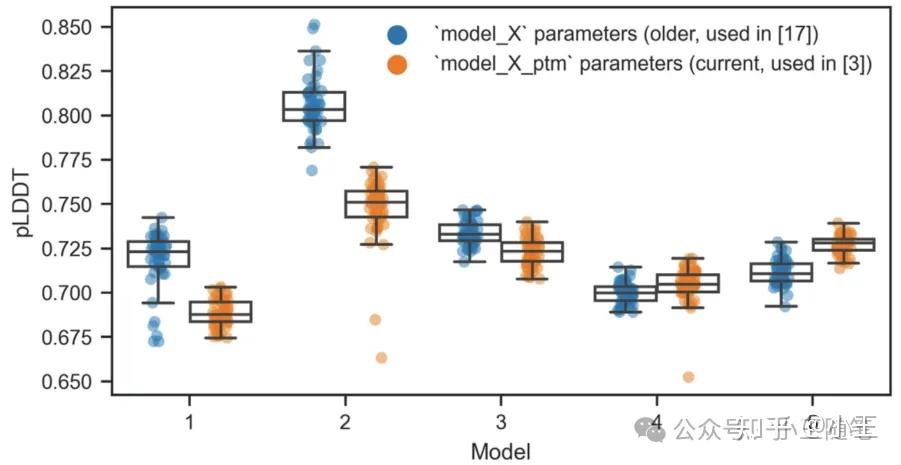
P2-4图B2。这里蓝色点对应的[17]指N3-1,橘色点对应的[3]指P1。
并且,Porter团队没有采用相同的采样条件来公平比较AF-Cluster和CF-Random。
这体现在采样。AF-Cluster的默认设置是MSA聚类之后,对每个序列聚类,仅使用AF2的5个模型中的1个预测1次——为了计算效率而牺牲采样。另一方面,CF-Random则会使用ColabFold的5个模型分别预测5次(5 models, 5 seeds),而后再取25个预测结果中pLDDT分值最高者为结果。那么,公平的比较显然应当是,或者AF-Cluster也使用5个模型分别预测5次,或者CF-Random也只用1个模型预测1次。
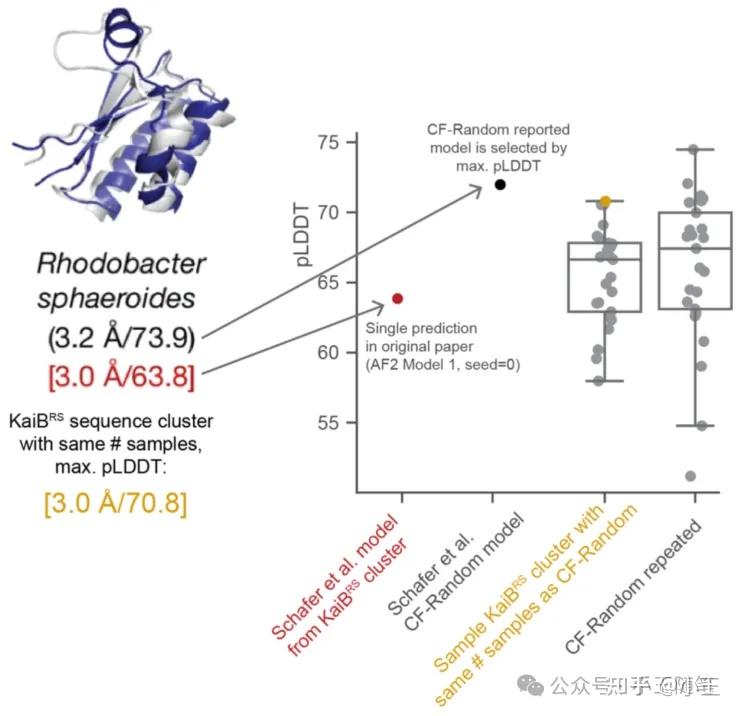
P2-4图3。
P2-4图3的左侧结构图来自N3-2论文图1b,我们在上方展示过,Porter团队意在通过这样的例子显示CF-Random的结果更好(pLDDT = 73.9),AF-Cluster的结果差(pLDDT = 63.8)——分别对应右侧图第1列红点和第2列黑点。然而,如果不计计算效率,令AF-Cluster有相同的采样次数(5个模型,5次预测)并取pLDDT最佳者,则其与CF-Random的结果大大接近(右侧图第3列黄点)。另一方面,如果不仅仅考虑pLDDT最佳者,而是考察所有采样结果,那么尽管CF-Random有更高的最高值,其均值不仅与AF-Cluster结果接近,而且有大得多的方差(右侧图第4列)。
简言之,如果不考虑计算成本,AF-Cluster和CF-Random的预测效果是很接近的,后者并没有如Porter团队所言远好于前者。
第四,针对N3-2,CF-Random不随机!Porter团队之所以取用CF-Random的名字,是因为他们随机采样MSA,构建浅深度的MSA作为ColabFold的输入。但是,P2-4指出,所谓“随机采样”并非随机。相反,CF-Random结合了两种特定的ColabFold设置,每种设置都各自独立地仅预测一种构象状态(P2-4图2A、C、D)。——这太扯了,基本上表明N3-2不应当通过同行评审发表在Nature。
P2-4图2。
最后,针对N1、N2-2、N3-2,Porter团队一再声称AF-Cluster方法成功预测折叠转换状态不是因为进化耦合。
P2-4做出了最有力的反驳:随机打乱MSA,从而彻底消融任何可能存在的进化耦合信号(图P2-4图4A),查看AF2的预测结果。符合预期,AF2的预测结果也是随机的(图P2-4图4B)。
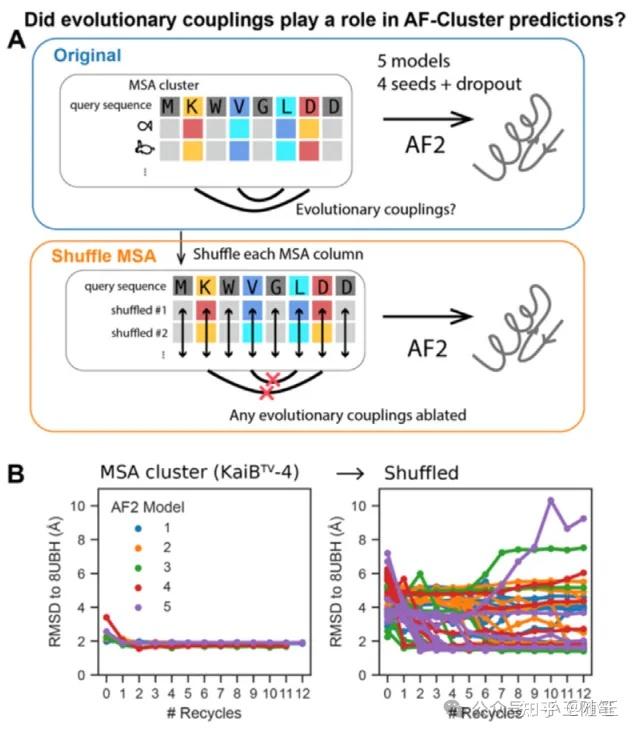
图P2-4图4A、B。
其实,让人很难想到的是,Porter团队居然会让自己的质疑滑向否定进化耦合。大概是他们在N2-2中针对Evoformer和recycling的消融实验(见上述第三回合),让其产生了“AF2是大记忆恢复术因而训练集中的结构模板会取代(override)进化耦合信号”的思想钢印吧。
还有若干细枝末节的反驳,我们限于篇幅不一一描述。
这样我们基本演绎完了五回合论战。我被Wayment-Steele这一方说服了,作为读者的你呢?
总结和思考1、明确研究目标、划定研究范围、区分研究对象是我们开展自己研究的首要步骤,也是我们学习和分析他人工作的首要步骤。
Porter团队针对AF-Cluster论文(P1)开展了极为敏捷的响应式研究(reactive research),在短短2个月时间内接连预印3篇完成度不一的论文,却始终混淆了AF-Cluster的研究目的,虚空打靶,以至于得到有违领域共识的结论——“AF2预测中进化耦合信号会被结构模板取代”。
这让我们不禁感慨,做研究有的时候还是要慢一点,再慢一点。在我还是本科交流学生时,我后来的博导Karl就告诫我,“两个月就能完成的几乎不可能是好的研究”(见《悼念我的导师Karl Freed》)。
另一方面,先构建尽可能全面的、大规模的基准测试集,再逐一测试各种工具方法,是机器学习/深度学习领域的研究范式。但是,如果不就事论事(case by case),细加辨别囊括入测试集的成员是否具备相当的共性,就很容易在测试后得出误导性的结论。针对折叠转换蛋白,我们需要辨别的是,给定一组折叠转换蛋白,它们之间构象转换的原因是什么?而不能如Porter课题组一样,囫囵吞枣,把成因不同的蛋白全部归入同一个基准测试集。
这也是为什么我在之前文章中说,“在学习和研究路径上,扎实的数学基础、物理图像、生化知识是做好基于深度学习的计算生物学的前提,而不是相反——在缺失理化知识时,从深度学习或人工智能出发,走向生物大分子的计算研究。”(见《随谈:我们能通过公众号读论文吗?》)
2、学术界,特指Nature出版社,的发表规范是否有待统一?
P1一篇发表在Nature的精彩的研究性论文,故而反驳P1的“matters arising”论文N3-2也可以发表在Nature。
但是,通过P2-4的逐条反驳,我们已经知道N3-2的方法和结论有着很容易看出来的很大问题——N3-2应该不能通过Nature的同行评审才对。
另一方面,反驳N3-2的论文P2-4,为什么不能再次发表在Nature,甚至不能发表在Nat Comms(如同N2-2),而只能发在JMB(影响因子 = 4.5)?这是否有违公平?这么做最大的问题在于,只读Nature的大同行,可能会因为N3-2而留下P1(AF-Cluster)错误的印象。
这种问题恐怕我们是不能去问Nature编辑的。
本文完。
2026.2.7于深圳
https://blog.sciencenet.cn/blog-3458695-1521498.html
上一篇:悼念我的导师Karl Freed
下一篇:7年前提出的概念终于被学术界接受了,慨之
