博文
牛津大学Compton教授—关于报道纳米材料电催化性能的思考
||
理不辨不明,事不鉴不清。电化学方向必读!
摘要:
在阅读越来越多电催化的文献时,我们对经常出现的电化学缺陷感到惊讶,这在某些情况下完全否定了研究工作的价值。特别是,我们被ACS Nano上的一篇社论(Voiry, Chhowalla et al., 2018, 12, 9635-9638)所激发,构思了这篇文章的主题,该社论为测量和报告新电催化剂活性的“最佳实践”提供了“指导”。至少从电化学的角度来看,需要提出相反的观点,因为所提供的建议在某些地方与传统观点不一致,或者更直白地说,是完全错误的!在下文中,我们不寻求提供另一系列“最佳实践指导方针”,也不是“一套材料表征的必要条件”——这很可能最终是IUPAC委员会应有的活动——而是纠正、详述和发展ACS Nano编委会所提供的讨论,我们认为补充是可取和有帮助。
六个主题:
1.标准、形式和平衡电势(Standard, formal and equilibrium potentials)
2. 我们应该如何量化电极动力学(electrode-kinetics)?
3. 什么是超电势?
4. 什么是起始电位?
5. 在半反应的电流-势图中,什么是合适的Tafel区域来分析“Tafel-slope”?
6. 单位和电化学表面积
正文:
在本文中,我们将关注六个与Voiry、Chhowalla等人在最近的ACS Nano中提出的建议相关的主题[1][2]。在每个部分中,我们首先做一个简短的陈述,我们认为这是正确的,但与Voiry,Chhowalla等人的陈述不同。在此简短陈述之后,对当前的问题进行了更深入的讨论和探讨。
1.标准、形式和平衡电势(Standard, formal and equilibrium potentials)
在大多数情况下,标准氧化还原电势不等于平衡电势。标准电势是在标准条件(STP)下定义的平衡电极电势,所有电活性物质均处于单位活度,气体处于标准压力(理想气体)。
以下定义对于ACS Nano [1]中的社论以及稍后讨论的方面都是至关重要的。为了保持清晰度,我们集中于以下简单的单电子转移过程的示例:
 对于该反应,电极电势(E)的能斯特方程为:
对于该反应,电极电势(E)的能斯特方程为:

其中ai是第i物种的活度,E⦵A/B是A/B氧化还原对的标准氧化还原电位。后一个值与反应的标准吉布斯能直接相关。如果我们在浓度基础上的用活度系数来表示这些电活性溶质的溶液相对活度,那么就可以用这个表达式: ai = γici/c⦵,这里c⦵是标准浓度(1 mol dm−3),可以得到方程(3)。

这里的γi 是活度系数,E⦵f,A/B是A/B氧化还原对的形式(条件)电势。

首先,形式电势不是单独依赖于A/B氧化还原对的固定值,而是特定于一组给定的实验条件。如果电活性物质的活度系数改变,形式电势也相应改变。对于单电子的情况,只有γB/γA = 1,标准电势等于形式电势。其次,上述的反应物和产物都是溶质,如果反应涉及到气体,那么标准电势和形式电势通常明显不同[4]。这是由于大多数非极性气体(如氢气和氧气)在水溶液中的溶解度较低[5]。因此,在一个大气压下,气体通常只存在于毫摩尔浓度的溶液中,相比之下,形式电势是在(可能的假设)单位浓度的基础上定义的[6]。因此,溶液相溶解气体的浓度在这两个定义之间通常有三个数量级的不同。
对于一个简单的单电子转移过程,我们可以将平衡电势定义为[7]:

即使对于这个简单的单电子转移反应,方程(5)对氧化(A)和还原(B)的比例也十分敏感。如果溶液中生成物的浓度比反应物的浓度高一个数量级,则体系的平衡电势会比形式电势减少~59.1 mV。这个问题似乎仍尚有争议,但在实际情况中如果一个电极反应涉及多个步骤,这些定义之间的差异变得更加明显。例如,如果反应中每个电子传递一个质子,则平衡电势随59.1 mV pH-1变化。在接近中性的pH值下进行的电催化实验往往与标准条件相差甚远!
2. 我们应该如何量化电极动力学(electrode-kinetics)?
对于半反应,交换电流(i0)的概念只在特定实际情况下是直接相关的,即当电活性产物和反应物都存在于溶液中,并且反应在一定程度上是可逆的。因此,我们不能用i0作为电催化活性的参数。
在电化学中,巴特勒-沃尔默方程(Butler-Volmer)有两种常用的公式。首先是物理电化学家使用的更普遍的形式,它接近于由Erdey-Gruz和Volmer推导出的第一个形式[8]:
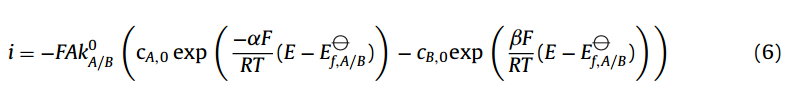
i为电流(A), k0为标准(严格意义上是形式)[9]电化学速率常数(m s−1),ci,0为第i种物质在电极表面的浓度。
第二种形式,源于Laidler等人的工作[10]。在材料和工程文献中经常使用,其表达式为:
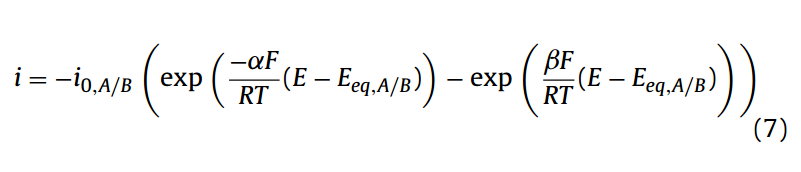
其中i0,A/B为A/B氧化还原对的交换电流(A)。两个表达式都假定α+β= 1。这两个方程((6),(7))之间的第一个主要区别是它们电势的参考,在第一种情况下,电势是相对于形式电势来测量的(式(4)),而第二种方法使用平衡电势(式(5))。Eq.(7)可以由Eq.(6)推导出来,通过重新整理平衡电势(Eq.(5))的定义,用平衡电势给出形式电势的定义,将这个定义代入Eq.(6)。进一步,对于式(7),假设表面浓度项(ci,0)等于在体相浓度。在平衡电势下,阳极电流和阴极电流大小相等,但方向相反,总和为零电流。在此电势下,阳极或阴极方向的电流为:

这种所谓的“交换电流”几乎没有物理意义,如Eq.(6 )所示,反应的速率是由k0A/B和α共同控制。
Eq.(7)的使用隐含了这样一种假设,即还原和氧化两种物质都存在在体相溶液中。例如经典的质子/氢氧化还原反应中[11],酸和溶解的氢气都存在溶液中,H+ + e− ⇄ 1/2H2。Voiry、Chhowalla等人强调了利用经典方法估算/测量交换电流(i0)来量化催化剂活性的重要性[12]。对于H+/H2反应,我们可以有效地定义和测量相关联的交换电流(i0)。但是,我们可以看到即使是对于上文给出的单电子例子,如果产物(cB)的浓度为零,那么平衡电势(Eq.(5))和交换电流(Eq.(8))都没有定义!
我们面临的问题是,如果只有反应物存在于体相溶液中,如何测量交换电流?这是一种非常常见的情况,例如,当研究氢气的析出反应或二氧化碳的还原时,通常氢气和二氧化碳还原过程的产物(甲酸盐、草酸盐、一氧化碳等)最初都不存在于体相溶液中。因此,任何通过对i0的测量试图为电催化材料性能的量化提供通用的指导方针的尝试都是没有帮助的,因为这个量(i0)的测量只在特定情况下才能成立。而对于独立的半反应 A + e− → B 或 A − e− → B,测量k0和α(或1-α)是优先考虑的。
我们注意到,在任何给定的电势下,微观可逆性的原理[13]要求阳极和阴极反应具有相同的过渡态,因此当以相同的电位测量时,对于一个单电子转移过程α+β= 1。然而,对于显著不可逆性电对,α和β的值通常是在在不同的电位分别评估阴极和阳极过程。由于微观可逆性的原理严格意义上只在相同电势下成立,当在不同电位下测量α和β相加不等于1。在其他电势下,α和β的相对大小可能随着过渡态变化而变化,例如,可能会因为溶剂化、吸附或双层结构随着施加的电位改变而改变。因此,通常实验测量的转移系数α+β≠1。因此,通过线性外推阴极和阳极的伏安图来给出Eeq和i0是错误的。
最后,对于多步反应来说,其机理以及相应的反应产物往往是与电位有相关性的。考虑两个平行发生的电极反应,一个例子可能是假设将CO2还原成CO或甲酸盐。例如,某些新型催化剂上CO的形成,其标准电化学速率常数(相对)较低但是传递系数很高;另一个反应,在这种情况下导致甲酸盐的形成,有一个高的标准电化学速率常数但传递系数低,那么电化学反应产物将有电位相关性。在低电位条件下,标准电化学速率常数较高的反应将占主导地位,在这个例子中,CO2电还原将生成甲酸盐。相反,在高过电位时,这两种过程的相对速率会发生转换,反应主产物将是CO。对于多步过程,电极反应机理的性质往往会随着外加电势的变化而变化!
3. 什么是过电势?
在溶液中只有电活性试剂存在的情况下,在稳态质量输运状态下,我们可以用伏安半波电势与可逆过程中预期的半波电势之差作为施加的过电势的度量。然而,在给定的一组实验条件下,尤其对于多步反应来说,精确计算这种预期的可逆半波电势并不一定是容易的[4]。
上一节所概述的有关交换电流的问题也引出了在IUPAC关于过电位的定义中存在的问题。IUPAC严格定义的过电势(ηe = E−Eeq)[7]是与平衡电势相关,但通过Eq.(5),如果cA或cB为零,则根据此定义不能定义过电势! 然而,这并不意味着这样一个系统的热力学也是不明确的,我们只是需要一个不同的定义。一般来说,如果一个电化学过程在一个给定的质量传输区域被描述为“可逆的”,这意味着电极表面还原和氧化物的浓度可以通过Nernst方程(即它们是局域平衡)很好的描述。
在文献中,我们很常见到在只有反应物在体相溶液中超电势甚至在某些情况下,标准的电势(ηs = E−E⦵),是在形式电势的基础上被定义的。对于一个简单的单电子转移过程,以Eq.(6)为启发,超电势的定义是明确和合理的。对于一个简单的单电子转移过程,超势的定义是明确和合理的,以Eq.(6)为灵感。例如,如果我们考虑可逆的二茂铁甲醇的单电子氧化反应,Fc − e− ⇄ Fc+,在稳态质量输运条件下,伏安半波电势近似等于其形式电势[14]。当还原态和氧化态的扩散系数相等时,可逆半波电势(E1/2)与体系的形式电势完全相等。对于析氢反应(H+ + e− ⇄ 1/2H2),由于其过程中涉及化学键的形成和破坏,情况则略有不同。在这里,我们只简要考虑在强酸水溶液中的析氢反应,并在该强酸水溶液中没有溶解的氢气。在此情况下,描述电极电势的Nernst方程可以表示为:
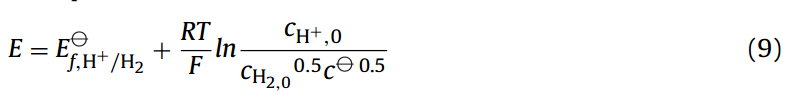
其中与氢浓度有关的平方根反映了反应的化学计量数。在稳态条件下,例如使用旋转圆盘电极,在伏安半波电势下,也就是50%的可用质子转化为氢的区域——并假设电活性物质的扩散系数相等——那么还原(氢)和氧化(质子)物质的表面浓度将等于:
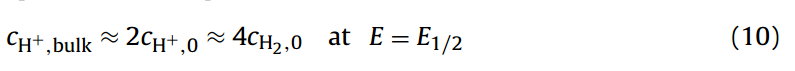
下标0表示物种的表面浓度。Eq.(10)表示在伏安半波电势下,质子(cH+,0)在电极表面浓度为其在体相溶液中(cH+,bulk)的一半。此外,根据反应化学计量学,在电极表面将一半的质子还原为氢气,意味着氢气在电极表面的浓度(cH2,0)等于在体相溶液中质子浓度的四分之一。Eq.10代入Eq.(9),得到伏安半波电势(E1/2)等于:
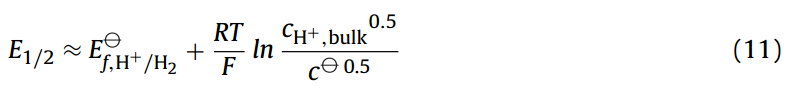
Eq.(11)只有在还原态和氧化态的扩散系数相等时才是完全正确的[15]。然而,重要的一点是,由于在电极表面的质量守恒和其化学计量反应,通过对伏安半波电势的测量得到的可逆伏安波的位置与反应的形式电势有关,并随这体相的酸浓度变化而变化[16]。
4. 什么是起始电位?
起始电位既不是热力学定义的参数,也不是动力学定义的参数;因此,这对于不同实验室(实验设置不同)之间电催化剂活性比较是没有用处的。一种可能更合适的方法,比如在测量ORR催化剂活性上用到的方法,是在给定的电位处报告法拉第电流密度[17]。
虽然“起始电位”是一个广泛报道的参数,乍一看似乎有着明确的定义[18]—例如“是在规定的条件下,在给定的电极上形成反应产物的最低过电位”或“是在观测到高于其背景电流的法拉第电流时的最低过电位”这些语句掩饰了问题的真实复杂性,下面将会解释。首先,考虑一个不可逆的单电子还原过程:
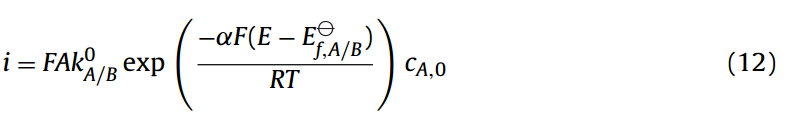
当E→+∞时,阴极电流趋近于零,当E→-∞时,阴极电流呈指数增长。要定义这样一个指数上升的“起点”必然需要一些额外的任意(arbitrary )定义。第二,对起始电位的一般解释是当还原或氧化的法拉第反应变得可测量的电位。很明显,这样的参数本质上是对体系的信/噪比(signal-to-noise)或信/背比(signal-to-background)的测量,因此它不仅反映了被研究材料的电催化性能,而且还受到测量条件的影响。为了举例说明这一点,图1(a)显示了在旋转圆盘电极上模拟的单电子不可逆还原反应。利用黑尔变换(Hale transform)[19]、[20],采用全隐式有限差分法对其伏安曲线进行了数值计算。理论预测的法拉第电流用黑色标出。图中也显示了一些电极其实际值的电容电流。特别是,在所施加的电极电势下,我们将电极电容近似为常数,一般金属在水溶液的电容值为20 μF cm−2 [21]。
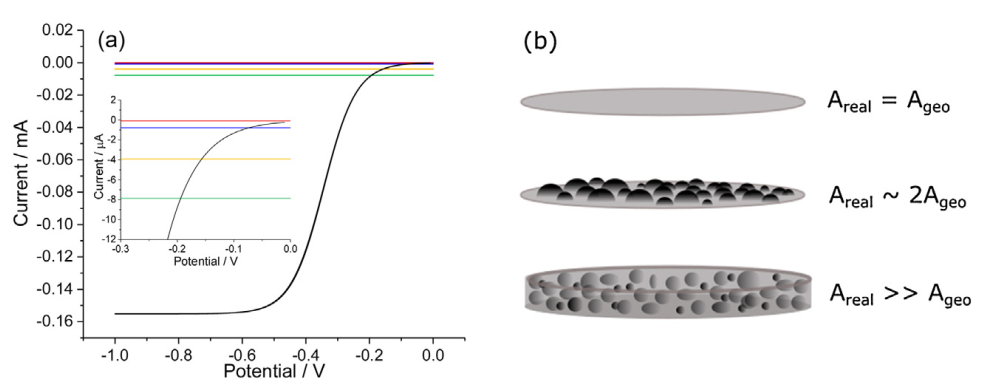
图1. (a)不同粗糙度因子(Rf) 下(Rf = 1(红色)、Rf = 10(蓝色)、Rf = 50(黄色)和Rf = 100 (绿色)),模拟5 mm直径的旋转圆盘电极上的法拉第电流(黑色)和电容电流的比较。内嵌图为电势范围在-0.3V至0V的放大版。 模拟所用参数: 扫速ν=0.02 V s-1, 旋转速度ω=1600 rpm,浓度= 1 mM, 扩散系数 D=1×10-9 m2 s-1, 形式电子转移速率常数 k0=1×10-7 m s-1,形式电势 Ef=0V,黏度=8.9×10-7 m2 s-1,转移系数α=β=0.5. 电容电流的计算公式为:Icap= Cdl(20 μF cm−2) × Rf× Ageo×ν.(b)不同粗糙度因素下的电极表面示意图。粗糙度因子Rf = 实际电极面积(Area)/几何面积(Ageo)。
为了计算出总的电极面积,我们将模拟的几何电极(Ageo/m2)面积乘以一个粗糙度因子Rf,即Areal = Ageo×Rf。正如后面所讨论的,对于非均相电极表面,另一个重要的参数是粗糙度因子Rf,其被用于计算催化剂的实际面积(Rf, catalyst)。如图1(b)所示,一个完美的原子级平整的电极的粗糙度因子为1。一个制备得很好的多晶电极的粗糙度系数为~ 1.5[17],而薄膜修饰的RDE的粗糙度系数在10-1000范围内。薄膜修饰的电极通常用于电催化剂实验[17],其中催化剂与非催化导电载体(如纳米碳)混合,然后作为薄层涂覆在电极表面。碳载体和催化剂的电容充电都会造成总背景电容电流。从Kocha等人提供的指南中可以看出,在他们实验的薄膜改性的直径5毫米的RDE的总背景电流为15 μA,这对应于一个有效的粗糙度因子约190。
从图1(a)中我们可以看到,如果我们在此模型情况下定义起始电位,当Faradaic电流的大小与电容的贡献相等时,发生此交叉的电位对电极的电容极为敏感。即使在这种理想化的情况下,这些实际电极电容的起始电位值变化超过200 mV。我们进一步建议将电容定义为一个常数是过度简化的,正如在原社论中所暗示的那样。
5. 在半反应的电流-电势图中,什么是合适的Tafel区域来分析“Tafel-slope”?
最好是定义一个相对于极限稳定状态值的电流范围,而不是定义一个适用于有意义的Tafel分析的电势范围,用以测量其传递系数,也可以给出相应的Tafel斜率。这里提供的分析表明,在提供一个适当的背景差来去除电容性电流的贡献后,只要进行了质量输运校正, 极限电流的10%到80%都适合用来进行动力学分析。如果忽略后一种校正,只有低于稳态电流20%的电流区间才适合用于动力学分析。
在Tafel图中,电流的对数log10|i|与外加电势E作图。如果我们假设反应是完全不可逆的和被Tafel方程很好的描述(Eq.(12)),那么我们可以看到对于单电子转移过程,这样一个半对数图的斜率等于−αF/(RT×ln10)和一个截距(在零超电势ηf)等于log10 (FAk0cA)。电催化实验最初是使用电流微扰技术的一种形式进行的,这种技术被称为整流器方法[23],[24]。在这里,一个预期的电流密度作用于工作电极上,cell随后被断开,相对于参照cell,快速改变测量的cell电位。通过测量断开cell的电势作为断开时间的函数,通过外推法得到原始cell的电势。因此,在电流/电位图表示的报告数据中,电位(测量变量)为y轴与电流(控制变量)为x轴被绘制在一起[24]。现代实验倾向于用电位计量法进行,因此人们可能会认为Tafel的坐标轴会被调换,但事实并非如此,积习难改( old habits die hard)。有关展现和分析Tafel图的其他方法的更多信息,请参见参考资料[25]。
根据Eq.(6),对于一个log10|i|对E的线性Tafel图,其要求该过程是完全不可逆的,且在整个电位分析范围内,电活性物质的表面浓度保持恒定。在实际中,通过扩散来补充活性物种是缓慢的,并且由于物质传输限制而产生扭曲。因此,有必要“纠正”在电极表面氧化还原活性物质的这些变化。该旋转圆盘电极是合理近似为uniformly accessible [26]、[27]。因此, 通过log10(1/I−1/Ilim)相对电势作图,在适当的扫描范围内考虑到了氧化还原活性物种表面浓度的变化。长期以来,人们一直主张进行传质修正[28]、[29]。
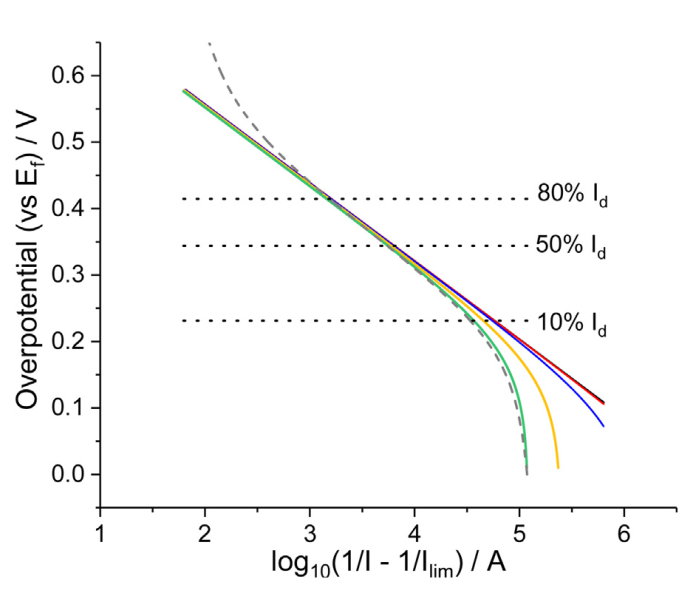
图2. 旋转圆盘电极的模拟传质校正Tafel图。内嵌图为电势范围在-0.3V至0V的放大版。 模拟所用参数: 扫速ν=0.02 V s-1, 旋转速度ω=1600 rpm,浓度= 1 mM, 扩散系数 D=1×10-9 m2 s-1, 形式电子转移速率常数 k0=1×10-7 m s-1,形式电势 Ef=0V,黏度=8.9×10-7 m2 s-1,转移系数α=β=0.5. 电容电流的计算公式为:Icap= Cdl(20 μF cm−2) × Rf× Ageo×ν,itot = ifarad + icap。
图2为一个模拟RDE实验的传质校正Tafel图。在这个模拟中,我们假设总电流可以简单地表示为法拉第和非法拉第贡献的总和(itot = ifarad + icap)。图2中的实线代表了该电极电容的贡献被假定为一个与电势无关的常数(icap = Cdl × Rf × Ageo × ν),虚线则假定该电极的电容随施加电势成线性变化icap = Cdl × Rf × Ageo × ν × f(E),函数f(E)是一个线性变化的无量纲标量,在模拟的伏安电位范围内,它在1和2之间呈线性变化。在没有背景校正来消除电容电流的情况下,图2显示了在电容背景电流的存在下,传质校正的Tafel图是如何被扭曲的。电容电流的产生是使用伏安技术的必然结果。这一结果首先强调了背景校正的重要性,获得消除非法拉第贡献的伏安数据。它也给出了一个明确的指示,即数据对修正的精度有多敏感。在此基础上,Mayrhofer等人先前提出,RDE伏安图的有益动力学校正是传质校正电流在稳态值的10%至80%之间[29],Kocha等人更加谨慎,并基于Vidal-Iglesias等人[31]建议,只有10-50%的电流区域是可用的。
6. 单位和电化学表面积
如果电子传递(而不是质量传递)是速控步骤,那么催化剂的电化学有效表面积是决定其催化能力的一个重要因素。两个化学上相关的量是粗糙度因子(催化剂表面积与电极几何面积之比的无量纲量)和电化学活性表面积(每克催化剂的催化活性表面积)。在实验上测量催化剂的实际活性表面积通常具有挑战性,在某些情况下对如何测量很敏感。
Voiry, Chhowalla等人[1]将电化学表面积(ECSA)和转化频率(TOF)作为无量纲参数呈现出来! 在指南中,ECSA被给出为特定双层电容(Cdl)相对于给定参考表面电容的无量纲比,这似乎遵循了ACS Catalysis一篇文章的定义[32]。类似地,TOF被给出为“nproduct/nsite”[1],如何定义和度量这两个量留给了读者。我们注意到,首先Voiry, Chhowalla等人将ECSA解释为粗糙因子,随后又混淆了文本中的术语。其次,我们认为表面积必须包含长度平方的单位,同样被称为“频率”的量的单位为时间的倒数。因此Voiry, Chhowalla等人所陈述的对于这些非常规的定义和一些派生的表达式应该需要重新考量。
在文献中,ECSA的定义可能因其使用的语境而异,然而在ORR领域,ECSA最有用的定义可能为每克物质(m2 gcatalyst- 1)的催化表面积[29]。这个值与催化剂的面积比活性(A mcatalyst- 2,每单位面积的催化法拉第电流[33])和质量比活性(A gcatalyst- 1,每单位质量的催化法拉第电流)有关。后两个值随电势的变化而变化,因此,为了获得相对活性的量度,这些值通常在给定的电位下报告。质量活性最直接的体现大多数是与铂基燃料电池的价格有关[34]。但从物理化学的角度来看,催化剂的比活性是一个重要的因素。催化剂的比活性的值可以用来衡量单位面积上的反应速率,但准确地确定比活性的大小并不容易,尤其是对于non-uniformly accessible的电极(多孔电极表面)来说。与这些值相关的是(无量纲的)催化剂粗糙度因子,该值是催化剂实际面积与电极几何面积之比。在某些文献中,粗糙度因子(如上文)也被定义为电极总电极表面与其几何面积之比。
催化剂表面积的测定不一定是直接的。请注意,ORR领域的“最佳实践”文档往往非常简短[17],提及的电极电容测量(或其他表面反应进程的测量,例如氢的欠电势沉积)需要使用模拟稳压器(analogue potentiostat)[29]。测量铂的电化学活性面积的主要错误可能是使用了阶梯扫描伏安法,几乎所有的现代商业稳压器把阶梯扫描伏安法作为了默认技术,这可能会导致低估铂的表面积,因此导致过高的估计材料的比活性。另外,蛋白质薄膜伏安法也会产生类似的问题[36]。阶梯伏安法的这个问题经常在恒电位器手册中提到[37],但是谁会阅读手册呢[38]?
本文所提及的一部分仅为观点,我们相信,在这个领域也会有其他人提出争议。该文也不是对Voiry, Chhowalla等人所写的社论的全部批判。可能本文中的一些陈述对于电化学领域的许多人来说是显而易见的,但我们希望对于非专业人士辨别了一些要点。
该文中所涉及的References与原文一致。
原文信息:
Li D, Batchelor-McAuley C, Compton R G. Some thoughts about reporting the electrocatalytic performance of nanomaterials. Applied Materials Today, 2020, 18: 100404.
链接:
https://www.sciencedirect.com/science/article/pii/S2352940719302926#eq0025
Applied Materials Today (2018-2019, IF= 8.013)介绍:
Applied Materials Today是爱思唯尔旗下材料学旗舰期刊系列 Materials Today 的重要成员。Applied Materials Today期刊创办于2015年,去年也是首年被SCI检索影响因子就突破8.0。期刊范围涵盖材料应用全领域研究,包括化学、物理、工程学和生物学,主要发表在新型电子、光学、机械和能源设备方面极具影响力的文章,同样也对医药领域、环境和社会有极大影响。
牛津大学 Richard G Compton教授介绍:
Compton教授于1977年在牛津大学(University of Oxford,英国)获得化学一等学士学位,于1980年在牛津大学获(师从W.J.Albery教授)得化学博士学位。
目前Compton教授就职于牛津大学物理化学系,从2011年至今被化学系授予Aldrichian Praelector头衔。Compton教授是国际著名的电分析化学专家,主要从事理论电化学、纳米电化学以及电化学传感技术研究,截止2020年3月,发表期刊论文1600篇以上(2001年至今,每年发表论文数超过50篇),总引用量(非自引)超过38000(H-index =102),从2014年至今,年年被Thomson Reuters评为‘highly cited researcher’。除外,Compton教授编写书籍7本,专利被应用在超过25个不同的领域,其中世界上第一支免校准pH计被PITTCON评为“最佳新产品”。
Compton教授是国际期刊《Electrochemistry Communications》(IF>4.8)的创刊人及主编,此杂志目前是电化学领域内影响因子最高的期刊之一,主要报道该领域内最新的研究进展。此外,Compton教授也是《Current Opinion in Electrochemistry》主编,还是另外10多个期刊的主编。
Compton教授究小组拥有较大的规模(约20人),设备齐全的现代化实验室,国际期刊Angewandte Chemie曾在2005年对Compton教授进行了采访,想更了解Compton教授可以看以下附图。
https://blog.sciencenet.cn/blog-3436572-1239263.html
上一篇:【Science综述】电催化理论与实验相结合:深入探究材料设计
下一篇:深切缅怀大化所催化科学家辛勤研究员