博文
Phenomics | 数据驱动的分枝杆菌表型组研究
|
The HumanPhenome Project近日,《表型组学》(Phenomics)杂志在线发表了中国科学院上海营养与健康研究所赵国屏院士、张国庆研究员题为“MPA: A Standardized Atlas for the Mycobacteriaceae Phenome Based on Heterogeneous Sources”的研究论文。
该研究采用数据驱动的方法建立了分枝杆菌科表型组的数据元素集,这是对现有微生物表型数据元素集的一个宝贵补充。通过使用分枝杆菌科表型组,该团队构建了最大、最完整的分枝杆菌科数据集,该数据集的应用将对分枝杆菌科的致病机制和抗菌研究提供新的视角。

扫二维码|查看原文
原文pdf链接:
https://link.springer.com/content/pdf/10.1007/s43657-023-00101-5.pdf
DOI:https://doi.org/10.1007/s43657-023-00101-5
引用格式:
Liu, W., Cen, H., Wu, Z. et al. Mycobacteriaceae Phenome Atlas (MPA): A Standardized Atlas for the Mycobacteriaceae Phenome Based on Heterogeneous Sources. Phenomics (2023). https://doi.org/10.1007/s43657-023-00101-5
研究背景
分枝杆菌耐药是非常严重的公共卫生问题,研究分枝杆菌科的表型及基因信息有助于致病性分枝杆菌临床感染的诊断和治疗。在微生物领域,多相分类信息被广泛用于识别微生物,而且毒力因子、抗菌素抗性、致病性等分子层面的信息也得到了越来越多的关注。随着可用于描述微生物的信息类型越来越多,仅使用多相分类法难以描述分子水平的表型,微生物表型本体(OMP)数据库等对微生物采集及生物特性的描述也不够。因此,该文研究者从宏观和微观的特征中收集了多尺度的表型,并将其整理为分枝杆菌科表型组的数据元素。然后,基于这些数据开发了分枝杆菌科表型图谱(MPA),审编并注释了分枝杆菌科236个种和18个亚种的10755个菌株的表型数据,还提供了一个友好的界面来搜索和比较分枝杆菌科菌株的综合表型,可以通过https://www.biosino.org/mpa/免费访问。
研究结果
该团队制定了82项微生物表型作为分枝杆菌科表型组的数据元素,如图1所示。多相分类表型包括74个表型,分为5个类别和20个子类别。功能性表型包括8个表型,包含三大类:基因相关表型、蛋白质相关表型和化合物相关表型。

图1 MPA中的数据元素集概要
基于分枝杆菌科的82个表型数据元素,他们通过文献收集、数据库整合和Traitar注释等方式,并结合生物信息方法,收集了超过9000万个多相分类表型和分子表型。在分枝杆菌科的五个属中,分枝杆菌属(Mycobacterium)包括最多的菌株(图2),有8083个。其中,6990个菌株属于结核分枝杆菌(M. tuberculosis)种,这可能是因为结核分枝杆菌是致病率最高的病原菌。拟分枝杆菌属(Mycobacteroides)包括第二多的测序菌株,这可能是由于该属与肺部、皮肤和软组织感染有关。
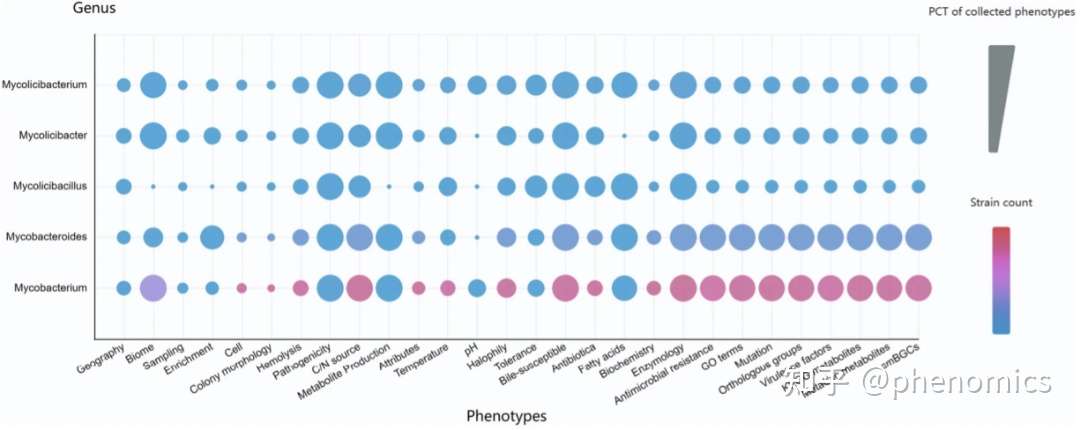
图2分枝杆菌科五个属的表型分布
他们统计了MPA中表型的完整性,不同表型的完整性差异很大,详细结果见图3。例如,毒力因子表型的完整性很高,可以达到93.54%,而霉菌酸模式的缺失则很明显,只有1.14%的菌株有记录。此外,基于菌株序列预测的多相表型的完整性较好,如抗生素物质、利用C/N源的能力和C/N源等。然而,超过三分之二的多相表型在MPA中的完整性低于50%,也难以预测这些缺失的表型信息,而这种多相表型信息大部分需要通过相关的实验研究获得。这也说明目前对分枝杆菌科表型的实验研究仍然不足。至于功能表型,由于基因组不完整、现有的分枝杆菌科功能表型注释参考数据库有限,并非所有的表型都能达到100%的表型完整性。

图3 MPA中的表型完整性分布
该团队还开发了访问界面(https://www.biosino.org/mpa/),以方便用户搜索和比较表型数据。用户可以通过简单或高级搜索来检索感兴趣的分枝杆菌科相关表型信息(图4)。简单搜索允许通过物种名称、基因组ID或化合物名称进行模糊查询,而高级搜索则提供大规模的复杂查询,包含五个多相模块(生态学、形态学、生理学、生物化学、酶学)和三个功能模块(基因相关表型、蛋白质相关表型、化合物相关表型)的23个术语。在结果页中列出了符合查询条件的菌株的概要信息,在每个菌株的详细页面中可以看到详细的表型信息。
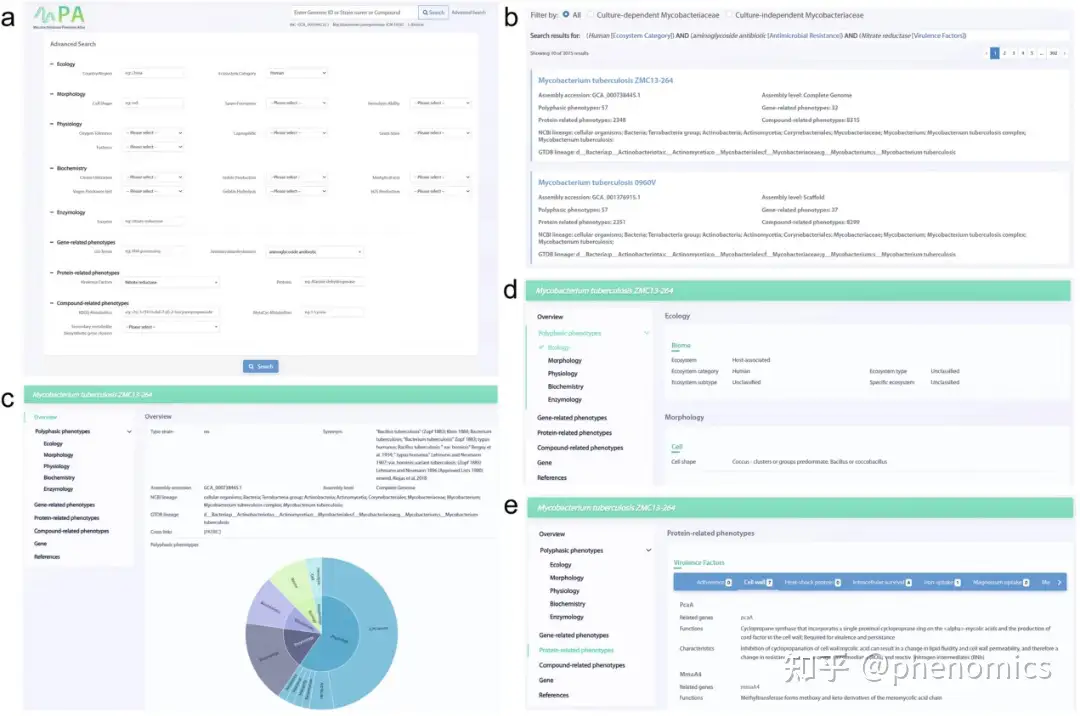
图4 MPA界面示例
用户可以通过表型比较功能一次比较多达4个菌株的41个表型。他们比较了Mycolicibacterium vanbaalenii DSM 7251和Mycolicibacterium mageritense DSM 44476之间的表型,并再现了文献中描述的两株菌的表型差异,还从分子角度发现了文献中没有收集到的差异性状,这将有助于研究人员找到研究课题的关键线索。另一方面,表型比较功能可以作为菌株进行初步分类和鉴定的有用工具。例如,表型比较可用于Mycolicibacterium chlorophenolicum DSM 43826的重分类(图5)。

图5用于重新分类Mycolicibacterium chlorophenolicum DSM 43826的表型比较结果
毒力因子基因对菌的毒力或致病性至关重要,并可能成为抗菌药物的靶点或帮助选择抗菌药物。结核分枝杆菌是一种已经广泛传播且未被完全控制的病原体。在151个毒力因子中,该文研究者发现36个毒力因子与结核分枝杆菌的基因组共同富集,表明这些毒力因子与结核分枝杆菌的毒力共同进化。例如,共同富集的ESX-3 T7SS分泌某些对铁吸收至关重要的效应物,而其他分泌的效应物则以不依赖铁的方式调节毒力(图6a,b)。结核分枝杆菌的共富集ESX-5分泌系统对细菌毒力和大型PE/PPE蛋白家族的分泌至关重要(图6a,c)。共同富集的硝酸盐还原酶有助于结核分枝杆菌在炎症或坏死组织的O2耗竭区生存(图6a,d)。MPA中致病性相关表型的拓扑数据分析有可能被用作研究结核分枝杆菌的致病性演变、其分子机制和抗菌靶点的有效工具。

图6 结核病的致病性相关表型的TDA网络富集模式
研究结论
该研究采用数据驱动的方法建立了分枝杆菌科表型组的数据元素,这是对现有微生物表型相关数据库的一个宝贵补充。通过使用分枝杆菌科表型组,他们构建了最大、最完整的分枝杆菌科数据集MPA。对MPA的拓扑数据分析揭示了结核分枝杆菌与毒力因子之间的共同进化,并发现了潜在的致病性相关表型。通过Fisher's精确检验,发现了260条潜在的病原体富集通路。MPA的应用可能会对分枝杆菌科的致病机制和抗菌研究提供新的视角。
中国科学院上海营养与健康研究所赵国屏院士、张国庆研究员为共同通讯作者,刘婉、岑卉和吴祉乐为共同第一作者。该项目获得国家重点研发计划(2021YFF0703702、2021YFC2301502和2018YFA0900704)、中国科学院战略优先研究计划(XDB38030100)、上海市科技重大专项(2017SHZDZX01)、中国科学院生物资源计划(KFJ-BRP-017-79和KFJ-BRP-009-001)的资助。
Abstract
The bacterial family Mycobacteriaceae includes pathogenic and nonpathogenic bacteria, and systematic research on their genome and phenome can give comprehensive perspectives for exploring their disease mechanism. In this study, the phenotypes of Mycobacteriaceae were inferred from available phenomic data, and 82 microbial phenotypic traits were recruited as data elements of the microbial phenome. This Mycobacteriaceae phenome contains five categories and 20 subcategories of polyphasic phenotypes, and three categories and eight subcategories of functional phenotypes, all of which are complementary to the existing data standards of microbial phenotypes. The phenomic data of Mycobacteriaceae strains were compiled by literature mining, third-party database integration, and bioinformatics annotation. The phenotypes were searchable and comparable from the website of the Mycobacteriaceae Phenome Atlas (MPA, https://www.biosino.org/mpa/). A topological data analysis of MPA revealed the co-evolution between Mycobacterium tuberculosis and virulence factors, and uncovered potential pathogenicity-associated phenotypes. Two hundred and sixty potential pathogen-enriched pathways were found by Fisher's exact test. The application of MPA may provide novel insights into the pathogenicity mechanism and antimicrobial targets of Mycobacteriaceae.
通讯作者赵国屏

赵国屏,分子微生物学家,中国科学院院士。现任中国科学院上海营养与健康研究所生物医学大数据中心首席科学家,国科大杭州高等研究生命与健康科学学院教授。长期从事微生物生理生化、代谢调控及酶作用机理的研究。参与组织中国人类基因组计划,建立基因组学研究平台;启动生物芯片、生物信息学、蛋白质组学研究工作。在微生物基因组、代谢酶乙酰化组、肠道微生物组研究方面,做出开创性工作。开拓系统合成生物学研究领域,在天然化合物人工细胞工厂合成、单染色体酵母构建与CRISPR-Dx体系创建方面作出基础性贡献。通讯作者张国庆

张国庆,研究员、博士生导师,现任中国科学院上海营养与健康研究所生物医学大数据中心副主任。主要研究方向是生物医学数据库与知识库,包括精准医学、自然及疾病人群队列、人类表型组、环境及病原及人体微生物组等领域的数据库和知识库的研发,致力于多维生命组学数据、文献数据、健康与医疗等真实世界数据的集成与管理,以及以知识库为代表的数据科学关键技术研究。在国家与地方多项重大重点项目中,承担大数据平台的研发任务。第一作者刘婉

刘婉,博士,中国科学院上海营养与健康研究所生物医学大数据中心工程师,主要研究方向为微生物相关数据库与数据仓库、合成生物学数据库与数据仓库。第一作者岑卉

岑卉,中国科学院上海营养与健康研究所博士研究生。主要研究方向为微生物耐药预测模型及耐药突变位点数据库的构建。第一作者吴祉乐

吴祉乐,工程师。主要研究方向为已知CRISPR-Cas系统模块识别及未知Cas蛋白发掘。
Phenomics期刊简介

Phenomics是一本新创的同行评审国际期刊,聚焦表型组学前沿研究,搭建全球表型组学领域专家交流的国际平台,推动该领域相关的理论创新和学科发展。
本期刊拥有强大的国际编委团队,复旦大学金力院士担任主编,美国系统生物学研究所Leroy Hood院士、澳大利亚莫道克大学Jeremy Nicholson院士、德国莱布尼兹环境医学研究所Jean Krutmann院士、复旦大学唐惠儒教授共同担任副主编,复旦大学丁琛教授担任执行主编,另有来自全球多国的三十多位著名科学家共同组成编委团队,以及四十多位青年科学家组成青年编委团队。
我们诚挚地邀请广大科研人员投稿!
Phenomics官网:https://www.springer.com/journal/43657
投稿链接:https://www.editorialmanager.com/pnmc/
编辑部邮箱:phenomics@ihup.org.cn、phenomics@fudan.edu.cn
欢迎关注Phenomics官方公众号

文章来源:人类表型组计划公众号
https://blog.sciencenet.cn/blog-3558836-1397599.html
上一篇:Phenomics | Phenomics期刊2023年第三期文章合集
下一篇:Phenomics | 沈侠团队揭示大脑基因异构体比例的遗传效应