博文
Phenomics | Phenomics期刊2023年第三期文章合集
|
本文介绍了Phenomics期刊2023年第三期收录文章合集,文章概览如下,请查收!

(Phenomics期刊2023年第三期封面图)
01
Hidden Genetic Regulation of Human Complex Traits via Brain Isoforms

irQTL结果汇总及示例

扫二维码|查看原文
DOI:
https://doi.org/10.1007/s43657-023-00100-6
引用格式:
Pan, L., Zheng, C., Yang, Z. et al. Hidden Genetic Regulation of Human Complex Traits via Brain Isoforms. Phenomics 3, 217–227 (2023). https://doi.org/10.1007/s43657-023-00100-6
该研究对大脑中特定的irQTL —— 控制基因的异构体表达相对比例而不是整体基因表达水平的数量性状位点进行了一系列研究。除了识别出数千个无 eQTL效应的 irQTL 外,作者还发现具有此类irQTL调控特性的基因富集某些人类复杂性状的遗传力,尤其是神经相关表型。通过孟德尔随机化分析,该研究衡量了受遗传调控的异构体比例对人类神经相关表型的下游因果效应。研究强调了量化和研究异构体表达对于探索人类复杂性状与疾病遗传机制的重要性。
Abstract
Alternative splicing exists in most multi-exonic genes, and exploring these complex alternative splicing events and their resultant isoform expressions is essential. However, it has become conventional that RNA sequencing results have often been summarized into gene-level expression counts mainly due to the multiple ambiguous mapping of reads at highly similar regions. Transcript-level quantification and interpretation are often overlooked, and biological interpretations are often deduced based on combined transcript information at the gene level. Here, for the most variable tissue of alternative splicing, the brain, we estimate isoform expressions in 1,191 samples collected by the Genotype-Tissue Expression (GTEx) Consortium using a powerful method that we previously developed. We perform genome-wide association scans on the isoform ratios per gene and identify isoform-ratio quantitative trait loci (irQTL), which could not be detected by studying gene-level expressions alone. By analyzing the genetic architecture of the irQTL, we show that isoform ratios regulate educational attainment via multiple tissues including the frontal cortex (BA9), cortex, cervical spinal cord, and hippocampus. These tissues are also associated with different neuro-related traits, including Alzheimer’s or dementia, mood swings, sleep duration, alcohol intake, intelligence, anxiety or depression, etc. Mendelian randomization (MR) analysis revealed 1,139 pairs of isoforms and neuro-related traits with plausible causal relationships, showing much stronger causal effects than on general diseases measured in the UK Biobank (UKB). Our results highlight essential transcript-level biomarkers in the human brain for neuro-related complex traits and diseases, which could be missed by merely investigating overall gene expressions.
02
Diets, Gut Microbiota and Metabolites
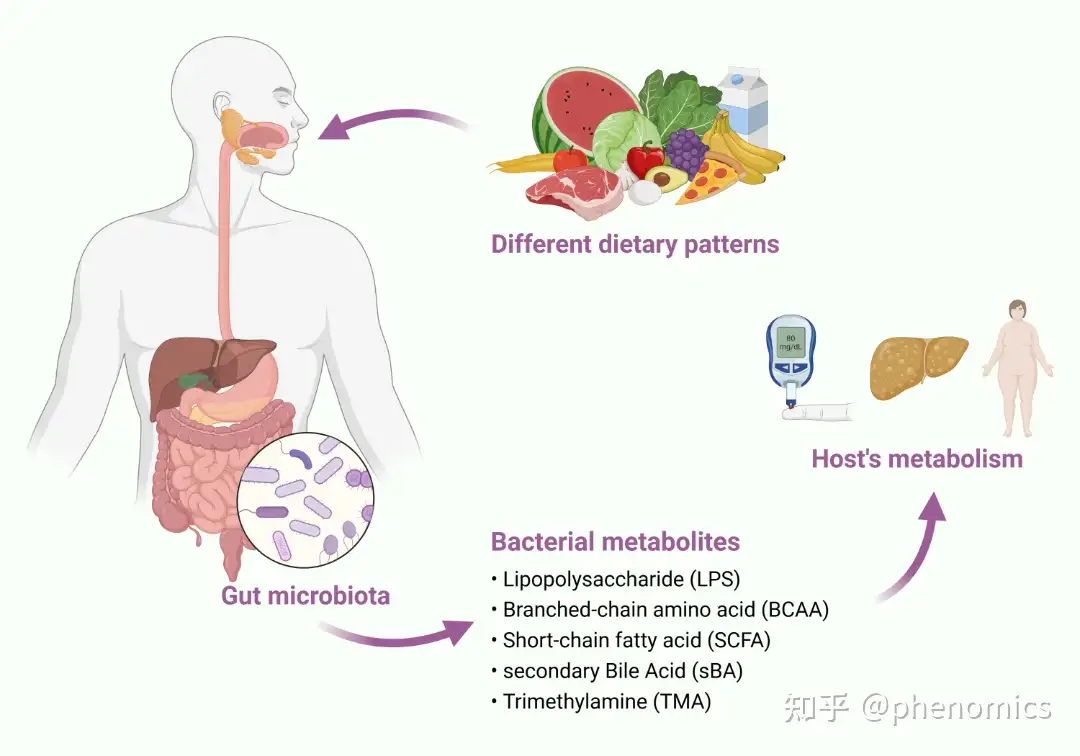
饮食、肠道菌群和代谢

扫二维码|查看原文
DOI:
https://doi.org/10.1007/s43657-023-00095-0
引用格式:
Liu, Y., Zhong, W., Li, X. et al. Diets, Gut Microbiota and Metabolites. Phenomics 3, 268–284 (2023). https://doi.org/10.1007/s43657-023-00095-0
肠道微生物群是指生活在肠道内的微生物的总集合,它们在营养物质的吸收和消化中起着至关重要的作用。在过去的几十年里,新一代“组学”(宏基因组学、转录组学、蛋白质组学和代谢组学)技术使精确识别微生物群和代谢物成为可能,还能描述它们在个体、群体甚至同一受试者的不同时间点之间的变异性。经过大量的努力,现在人们普遍认为肠道微生物群是一个动态变化的种群,其组成受到宿主健康状况和生活方式的影响。饮食是塑造肠道菌群的主要因素之一。饮食的成分因国家、宗教和人口的不同而不同。一些特殊的饮食已经被人们采用了几百年,而其潜在的机制在很大程度上仍然未知。最近的研究表明,饮食可以迅速地改变肠道微生物群。饮食中的营养物质及肠道微生物群产生代谢物的独特模式与疾病的发生有关,包括肥胖、糖尿病、非酒精性脂肪性肝病、心血管疾病、神经疾病等。该文综述了不同饮食模式对肠道菌群组成、细菌代谢物及其对宿主代谢的影响的最新进展。
Abstract
The gut microbiota refers to the gross collection of microorganisms, estimated trillions of them, which reside within the gut and play crucial roles in the absorption and digestion of dietary nutrients. In the past decades, the new generation ‘omics’ (metagenomics, transcriptomics, proteomics, and metabolomics) technologies made it possible to precisely identify microbiota and metabolites and describe their variability between individuals, populations and even different time points within the same subjects. With massive efforts made, it is now generally accepted that the gut microbiota is a dynamically changing population, whose composition is influenced by the hosts’ health conditions and lifestyles. Diet is one of the major contributors to shaping the gut microbiota. The components in the diets vary in different countries, religions, and populations. Some special diets have been adopted by people for hundreds of years aiming for better health, while the underlying mechanisms remain largely unknown. Recent studies based on volunteers or diet-treated animals demonstrated that diets can greatly and rapidly change the gut microbiota. The unique pattern of the nutrients from the diets and their metabolites produced by the gut microbiota has been linked with the occurrence of diseases, including obesity, diabetes, nonalcoholic fatty liver disease, cardiovascular disease, neural diseases, and more. This review will summarize the recent progress and current understanding of the effects of different dietary patterns on the composition of gut microbiota, bacterial metabolites, and their effects on the host's metabolism.
03Sample Collection, DNA Extraction, and Library Construction Protocols of the Human Microbiome Studies in the International Human Phenome Project
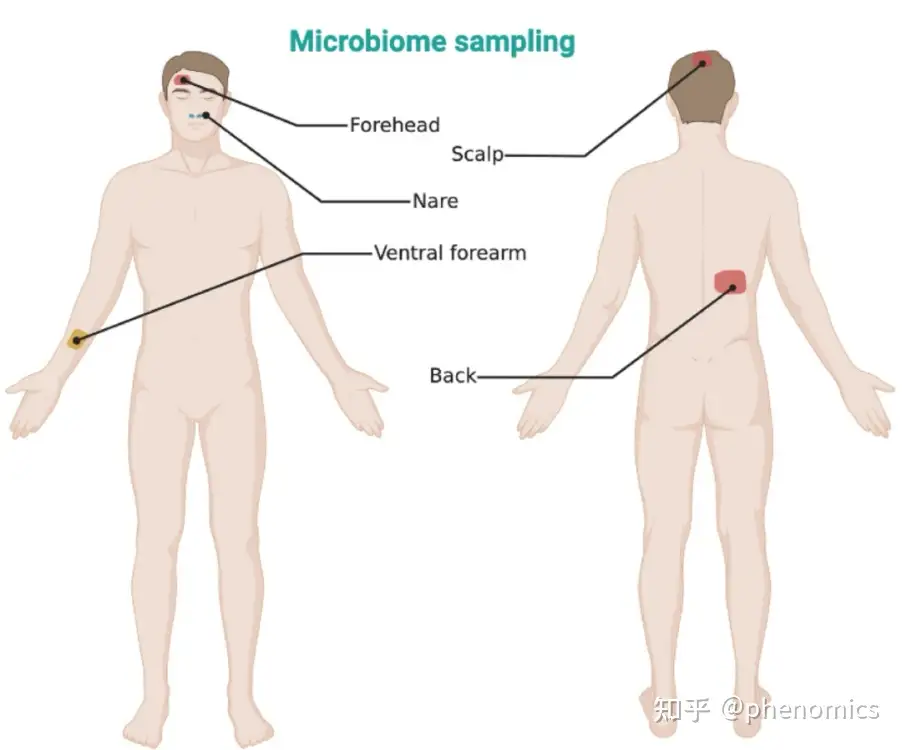
鼻腔和皮肤采样部位示意图

扫二维码|查看原文
DOI:
https://doi.org/10.1007/s43657-023-00097-y
引用格式:
Wang, Y., Zhang, R., Pu, Y. et al. Sample Collection, DNA Extraction, and Library Construction Protocols of the Human Microbiome Studies in the International Human Phenome Project. Phenomics 3, 300–308 (2023). https://doi.org/10.1007/s43657-023-00097-y
该研究提供了针对包括鼻腔、口腔、皮肤以及粪便在内的人体微生物样本的样本采集、DNA提取和文库构建的详细操作方法,为人体微生物组研究提供了一套可重复的样品处理标准流程。
Abstract
The human microbiome plays a crucial role in human health. In the past decade, advances in high-throughput sequencing technologies and analytical software have significantly improved our knowledge of the human microbiome. However, most studies concerning the human microbiome did not provide repeatable protocols to guide the sample collection, handling, and processing procedures, which impedes obtaining valid and timely microbial taxonomic and functional results. This protocol provides detailed operation methods of human microbial sample collection, DNA extraction, and library construction for both the amplicon sequencing-based measurements of the microbial samples from the human nasal cavity, oral cavity, and skin, as well as the shotgun metagenomic sequencing-based measurements of the human stool samples among adult participants. This study intends to develop practical procedure standards to improve the reproducibility of microbiota profiling of human samples.
04
Deep Immunophenotyping of Human Whole Blood by Standardized Multi-parametric Flow Cytometry Analyses
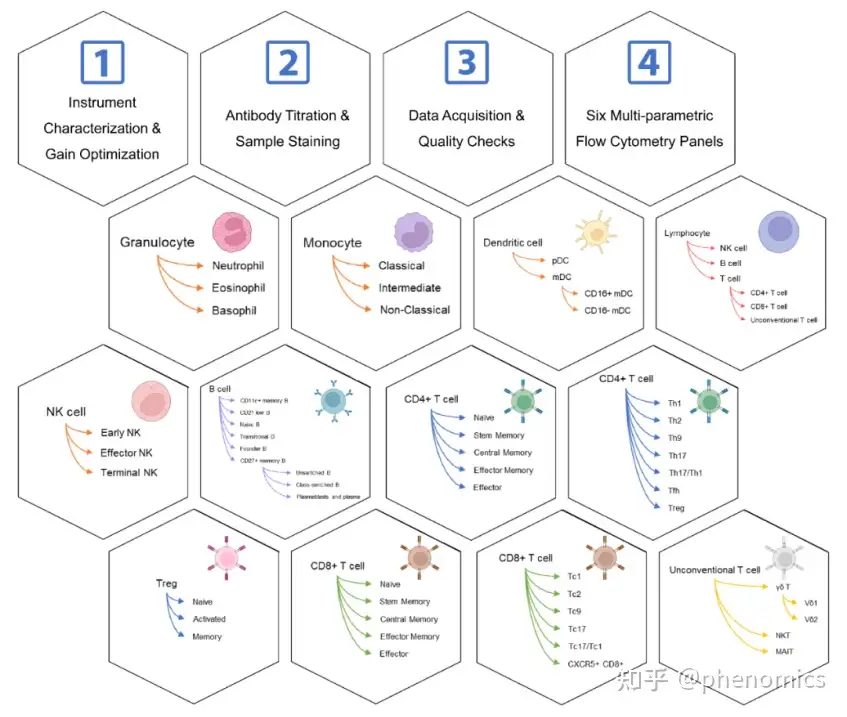
构建数个全新的11色多色流式细胞术Panel,对人体外周血主要免疫细胞亚群进行精细分型,实现人体免疫表型的深度刻画

扫二维码|查看原文
DOI:
https://doi.org/10.1007/s43657-022-00092-9
引用格式:
Gao, J., Luo, Y., Li, H. et al. Deep Immunophenotyping of Human Whole Blood by Standardized Multi-parametric Flow Cytometry Analyses. Phenomics 3, 309–328 (2023). https://doi.org/10.1007/s43657-022-00092-9
该研究设计了数个全新的、经过系统优化的11色流式细胞术抗体组合(Panels),对人体全血免疫细胞表型进行深度分析。该多色流式细胞术方案共包含51种经过验证的流式细胞术抗体,包括识别不同的免疫细胞亚群的细胞谱系标志物抗体,以及在鉴定免疫细胞亚群的同时评估其功能状态的细胞表面功能标志物抗体。为了确保数据的可靠性,该研究提供了一整套详细的标准操作程序,包括1) 流式细胞仪参数优化,2) 抗体滴定和样品染色,3) 数据采集和质量检查。该套标准化操作体系具有良好的实用性,已被应用于多个队列研究中。该方案中的流式多色Panel在探索性研究中也可自由组合及拓展,有助于更好地研究人类免疫系统的复杂性。
Abstract
Immunophenotyping is proving crucial to understanding the role of the immune system in health and disease. High-throughput flow cytometry has been used extensively to reveal changes in immune cell composition and function at the single-cell level. Here, we describe six optimized 11-color flow cytometry panels for deep immunophenotyping of human whole blood. A total of 51 surface antibodies, which are readily available and validated, were selected to identify the key immune cell populations and evaluate their functional state in a single assay. The gating strategies for effective flow cytometry data analysis are included in the protocol. To ensure data reproducibility, we provide detailed procedures in three parts, including (1) instrument characterization and detector gain optimization, (2) antibody titration and sample staining, and (3) data acquisition and quality checks. This standardized approach has been applied to a variety of donors for a better understanding of the complexity of the human immune system.
05
Deep Learning-Assisted Quantitative Susceptibility Mapping as a Tool for Grading and Molecular Subtyping of Gliomas
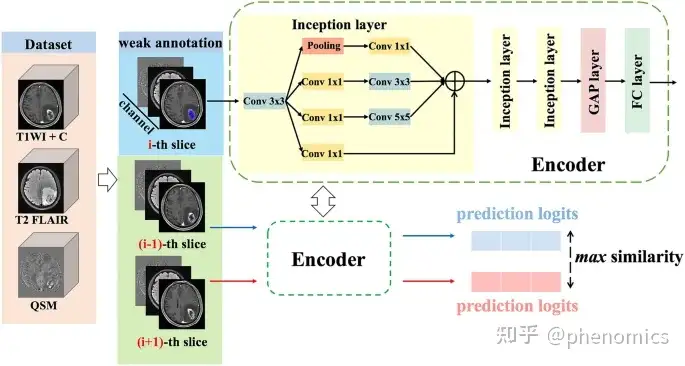
管道的初始卷积神经网络:对于多模态集成,将同一主体的图像切片在通道维度上进行拼接

扫二维码|查看原文
DOI:
https://doi.org/10.1007/s43657-022-00087-6
引用格式:
Rui, W., Zhang, S., Shi, H. et al. Deep Learning-Assisted Quantitative Susceptibility Mapping as a Tool for Grading and Molecular Subtyping of Gliomas. Phenomics 3, 243–254 (2023). https://doi.org/10.1007/s43657-022-00087-6
该研究旨在探讨深度学习(DL)辅助的定量易感性图谱(QSM)在胶质瘤分级和分子分型中的价值。该研究纳入42例胶质瘤患者,术前接受T2液体衰减反转恢复(T2 FLAIR)、对比增强t1加权成像(T1WI + C)和3.0T磁共振成像(MRI) QSM扫描。组织病理学和免疫组织化学染色用于确定胶质瘤分级、异柠檬酸脱氢酶(IDH) 1和α地中海贫血/智力迟钝综合征X连锁基因(ATRX)亚型;使用Insight Toolkit-SNAP程序(http://www.itksnap.org)手动进行肿瘤分割;采用初始卷积神经网络(CNN)和后续线性层作为训练编码器,从MRI切片中捕获多尺度特征;采用五重交叉验证作为训练策略(每重7个样本),训练、验证和测试数据集的样本量之比为4:1:1,以准确度和曲线下面积(AUC)作为评价指标;采用初始CNN,单模态QSM在鉴别胶质母细胞瘤(GBM)和其他级别胶质瘤(OGG, II-III级),并且在预测IDH1突变和ATRX丢失(准确率分别为0.80、0.77、0.60)方面优于T2 FLAIR(0.69、0.57、0.54)或T1WI + C(0.74、0.57、0.46)。与任何单一模式相比,三种模式联合使用时,在胶质瘤分级(OGG和GBM: 0.91/0.89/0.87,低级别和高级别胶质瘤:0.83/0.86/0.81)、预测IDH1突变(0.88/0.89/0.85)和预测ATRX损失(0.78/0.71/0.67)方面均达到最佳AUC/准确度/ F1评分。作为常规MRI的补充,DL辅助QSM是一种很有前途的分子成像方法,可用于评估胶质瘤分级、IDH1突变和ATRX丢失。
Abstract
This study aimed to explore the value of deep learning (DL)-assisted quantitative susceptibility mapping (QSM) in glioma grading and molecular subtyping. Forty-two patients with gliomas, who underwent preoperative T2 fluid-attenuated inversion recovery (T2 FLAIR), contrast-enhanced T1-weighted imaging (T1WI + C), and QSM scanning at 3.0T magnetic resonance imaging (MRI) were included in this study. Histopathology and immunohistochemistry staining were used to determine glioma grades, and isocitrate dehydrogenase (IDH) 1 and alpha thalassemia/mental retardation syndrome X-linked gene (ATRX) subtypes. Tumor segmentation was performed manually using Insight Toolkit-SNAP program (http://www.itksnap.org). An inception convolutional neural network (CNN) with a subsequent linear layer was employed as the training encoder to capture multi-scale features from MRI slices. Fivefold cross-validation was utilized as the training strategy (seven samples for each fold), and the ratio of sample size of the training, validation, and test dataset was 4:1:1. The performance was evaluated by the accuracy and area under the curve (AUC). With the inception CNN, single modal of QSM showed better performance in differentiating glioblastomas (GBM) and other grade gliomas (OGG, grade II–III), and predicting IDH1 mutation and ATRX loss (accuracy: 0.80, 0.77, 0.60) than either T2 FLAIR (0.69, 0.57, 0.54) or T1WI + C (0.74, 0.57, 0.46). When combining three modalities, compared with any single modality, the best AUC/accuracy/F1-scores were reached in grading gliomas (OGG and GBM: 0.91/0.89/0.87, low-grade and high-grade gliomas: 0.83/0.86/0.81), predicting IDH1 mutation (0.88/0.89/0.85), and predicting ATRX loss (0.78/0.71/0.67). As a supplement to conventional MRI, DL-assisted QSM is a promising molecular imaging method to evaluate glioma grades, IDH1 mutation, and ATRX loss.
06
Phenomic Studies on Diseases: Potential and Challenges

表型组学研究面临的主要科学问题和技术问题

扫二维码|查看原文
DOI:
https://doi.org/10.1007/s43657-022-00089-4
引用格式:
Ying, W. Phenomic Studies on Diseases: Potential and Challenges. Phenomics 3, 285–299 (2023). https://doi.org/10.1007/s43657-022-00089-4
全球的很多国家正在经历快速的人口老化。老化是多种重大疾病的关键风险因素,并且正在造成老化相关疾病患者人数的显著增加。为了克服这一严峻挑战,迫切需要建立起具有创新性的预防策略、诊断方法及治疗方法。不断增加的证据表明,人类表型组学为发现新的疾病风险因素、诊断标志物和精准疗法提供了颠覆性战略和方法。
Abstract
The rapid development of such research field as multi-omics and artificial intelligence (AI) has made it possible to acquire and analyze the multi-dimensional big data of human phenomes. Increasing evidence has indicated that phenomics can provide a revolutionary strategy and approach for discovering new risk factors, diagnostic biomarkers and precision therapies of diseases, which holds profound advantages over conventional approaches for realizing precision medicine: first, the big data of patients' phenomes can provide remarkably richer information than that of the genomes; second, phenomic studies on diseases may expose the correlations among cross-scale and multi-dimensional phenomic parameters as well as the mechanisms underlying the correlations; and third, phenomics-based studies are big data-driven studies, which can significantly enhance the possibility and efficiency for generating novel discoveries. However, phenomic studies on human diseases are still in early developmental stage, which are facing multiple major challenges and tasks: first, there is significant deficiency in analytical and modeling approaches for analyzing the multi-dimensional data of human phenomes; second, it is crucial to establish universal standards for acquirement and management of phenomic data of patients; third, new methods and devices for acquirement of phenomic data of patients under clinical settings should be developed; fourth, it is of significance to establish the regulatory and ethical guidelines for phenomic studies on diseases; and fifth, it is important to develop effective international cooperation. It is expected that phenomic studies on diseases would profoundly and comprehensively enhance our capacity in prevention, diagnosis and treatment of diseases.
07
A Genome-Wide Association Study for Susceptibility to Axial Length in Highly Myopic Eyes
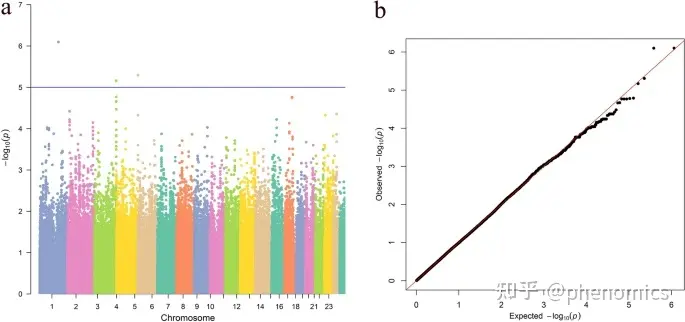
轴长全基因组关联研究(GWAS)的Manhattan和分位数图

扫二维码|查看原文
DOI:
https://doi.org/10.1007/s43657-022-00082-x
引用格式:
Lu, Q., Du, Y., Zhang, Y. et al. A Genome-Wide Association Study for Susceptibility to Axial Length in Highly Myopic Eyes. Phenomics 3, 255–267 (2023). https://doi.org/10.1007/s43657-022-00082-x
高度近视长期以来在世界范围内高度流行,而其中的遗传因素尚未得到较全面的解释。为了确定高度近视眼轴长(AL)的新易感基因,该文研究者使用来自350个高度近视患者的深度全基因组测序数据进行了全基因组关联研究(GWAS),他们鉴定了4个顶端SNP,发现ADAM金属肽酶With Thrombospondin Type 1 Motif 16 (ADAMTS16)和磷脂酰肌醇聚糖锚定生物合成类Z (PIGZ)具有潜在的临床意义。动物实验证实,在形态缺失小鼠中,特别是在神经节细胞层,可以观察到PIGZ的表达,且表达水平较高。ADAMTS16和PIGZ的信使RNA (mRNA)水平在失形眼的神经视网膜中均显著升高(p = 0.005和0.007),两种蛋白在失形眼的神经视网膜中均有显著的表达上调(p = 0.004和0.042)。富集分析揭示了细胞粘附和信号转导在AL中的重要作用,并且发现了几种与AL相关的通路,包括昼夜节律夹带和炎症介质调节瞬时受体电位通道。该研究在高度近视眼中发现了4个与AL相关的新SNP,并证实了ADAMTS16和PIGZ在剥夺眼的神经视网膜中表达显著上调。此外,富集分析为高度近视的病因提供了新的见解,并为未来的研究方向开辟了道路。
Abstract
High myopia has long been highly prevalent worldwide with a largely yet unexplained genetic contribution. To identify novel susceptibility genes for axial length (AL) in highly myopic eyes, a genome-wide association study (GWAS) was performed using the genomic dataset of 350 deep whole-genome sequencing data from highly myopic patients. Top single nucleotide polymorphisms (SNPs) were functionally annotated. Immunofluorescence staining, quantitative polymerase chain reaction, and western blot were performed using neural retina of form-deprived myopic mice. Enrichment analyses were further performed. We identified the four top SNPs and found that ADAM Metallopeptidase With Thrombospondin Type 1 Motif 16 (ADAMTS16) and Phosphatidylinositol Glycan Anchor Biosynthesis Class Z (PIGZ) had the potential of clinical significance. Animal experiments confirmed that PIGZ expression could be observed and showed higher expression level in form-deprived mice, especially in the ganglion cell layer. The messenger RNA (mRNA) levels of both ADAMTS16 and PIGZ were significantly higher in the neural retina of form-deprived eyes (p = 0.005 and 0.007 respectively), and both proteins showed significantly upregulated expression in the neural retina of deprived eyes (p = 0.004 and 0.042, respectively). Enrichment analysis revealed a significant role of cellular adhesion and signal transduction in AL, and also several AL-related pathways including circadian entrainment and inflammatory mediator regulation of transient receptor potential channels were proposed. In conclusion, the current study identified four novel SNPs associated with AL in highly myopic eyes and confirmed that the expression of ADAMTS16 and PIGZ was significantly upregulated in neural retina of deprived eyes. Enrichment analyses provided novel insight into the etiology of high myopia and opened avenues for future research interest.
08 RNA-Sequencing Reveals Gene Expression and Pathway Signatures in Umbilical Cord Blood Affected by Birth Delivery Mode

验证差异表达基因(DEGs)

扫二维码|查看原文
DOI:
https://doi.org/10.1007/s43657-022-00086-7
引用格式:
Liu, Y., Sun, K., Gan, Y. et al. RNA-Sequencing Reveals Gene Expression and Pathway Signatures in Umbilical Cord Blood Affected by Birth Delivery Mode. Phenomics 3, 228–242 (2023).https://doi.org/10.1007/s43657-022-00086-7
剖宫产(CS)会增加后代患I型糖尿病、哮喘、炎症性肠病、乳糜泻、超重和肥胖等的风险,然而其潜在机制尚不清楚。为了研究CS对脐带血基因表达的影响,该文研究者对8名选择CS出生的足月婴儿和8名匹配的顺产(VD)婴儿进行了RNA测序,随后进行了单基因分析、基因集富集分析、基因共表达网络分析和相互作用基因/蛋白分析。在另外20名CS和20名VD婴儿中进一步验证了上述鉴定的关键基因。他们首次发现,CS对免疫应答相关基因(IL12A、INFG、IL1B、TNF、MIF、IL4、CA1、if27、HLA-DOB、EPHB1)和代谢相关基因(DLK1、CYP2A6、GATM)的mRNA表达有显著影响。值得注意的是,与VD婴儿相比,CS婴儿血清TNF-α和IFN-γ显著上调(p分别为5.0 × 10-4和3.0 × 10-3)。CS可能通过影响上述过程中基因的表达而对后代健康产生不利影响。这些发现将有助于了解CS对健康不良影响的潜在潜在机制,并确定不同分娩方式出生的后代未来健康的生物标志物。
Abstract
Cesarean section (CS) confers increased risk of type I diabetes, asthma, inflammatory bowel disease, celiac disease, overweight and obesity, etc., in the offspring. However, the underlying mechanism remains unknown. To investigate the influence of CS on gene expression in cord blood, we have performed RNA-sequencing followed by single-gene analysis, gene set enrichment analysis, gene co-expression network analysis, and interacting genes/proteins analysis in eight full-term infants born by elective CS and eight matched vaginally delivered (VD) infants. Crucial genes identified above were further validated in another 20 CS and 20 VD infants. We found for the first time that mRNA expression of genes involved in immune response (IL12A, INFG, IL1B, TNF, MIF, IL4, CA1, IFI27, HLA-DOB and EPHB1) and metabolism (DLK1, CYP2A6 and GATM) were significantly influenced by CS. Notably, serum TNF-α and IFN-γ were remarkably up-regulated in the CS infants (p = 5.0 × 10–4 and 3.0 × 10–3, respectively) compared to the VD infants. It is biologically plausible that CS may exert adverse impacts on offspring health through influencing expression of genes in the above processes. These findings will help understand the potential underlying mechanisms of the adverse health impacts of CS and identify biomarkers for future health of offspring born with different delivery modes.
Phenomics期刊简介

Phenomics是一本新创的同行评审国际期刊,聚焦表型组学前沿研究,搭建全球表型组学领域专家交流的国际平台,推动该领域相关的理论创新和学科发展。
本期刊拥有强大的国际编委团队,复旦大学金力院士担任主编,美国系统生物学研究所Leroy Hood院士、澳大利亚莫道克大学Jeremy Nicholson院士、德国莱布尼兹环境医学研究所Jean Krutmann院士、复旦大学唐惠儒教授共同担任副主编,复旦大学丁琛教授担任执行主编,另有来自全球多国的三十多位著名科学家共同组成编委团队,以及四十多位青年科学家组成青年编委团队。
我们诚挚地邀请广大科研人员投稿!
Phenomics官网:https://www.springer.com/journal/43657
投稿链接:https://www.editorialmanager.com/pnmc/
编辑部邮箱:phenomics@ihup.org.cn、phenomics@fudan.edu.cn
欢迎关注Phenomics官方公众号

文章来源:人类表型组计划公众号
https://blog.sciencenet.cn/blog-3558836-1397598.html
上一篇:Phenomics | 盐胁迫下马齿苋幼苗如何生存? 巴西拉夫拉斯联邦大学使用多组学分析给出答案!
下一篇:Phenomics | 数据驱动的分枝杆菌表型组研究