博文
POWSC:scRNA-Seq功效分析工具
|||
论文标题:Simulation, Power Evaluation, and Sample Size Recommendation for Single Cell RNA-seq
杂志:Bioinformatics
作者信息:

软件安装:https://github.com/suke18/POWSC
输入数据:一个初始的scRNA-Seq数据集。
操作步骤:
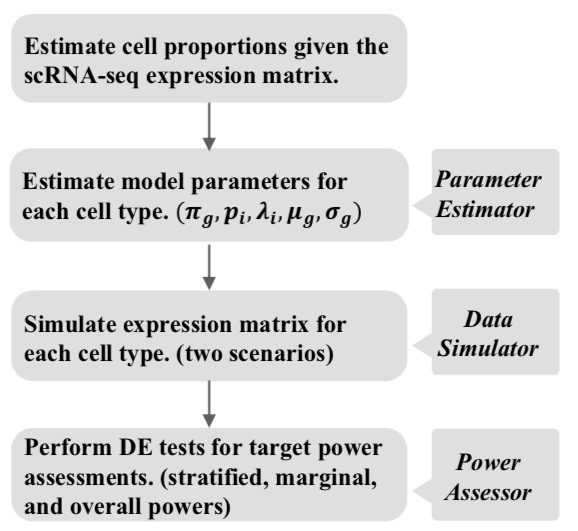
对scRNA-Seq数据进行降维和细胞聚类。这一步的目的是为了提高scRNA-Seq内部参数估计的准确性。如果对细胞不加分类直接估计参数,得到的参数不足以描述数据内部的结构。
对每一个细胞类,分别估计其数据分布的参数。这里我们用零膨胀珀松分布和对数式珀松分布的混合概率分布来表示每个类内部的数据分布。其中零膨胀分布表示的是抑制状态下的基因表达;对数珀松分布表示的是
基于概率分布生成模拟的差异表达(Differentially expressed,DE)数据。我们针对两种不同类型的差异表达数据进行模拟。
第一种是同一种实验条件下两个细胞类型之间的差异表达基因。这种情形比较简单,我们对每个细胞类模拟出基因的表达数据。至于差异表达基因,我们需要考虑两种情形:(i) 基因在两个细胞类型下分别是抑制和表达的。这就需要我们分别用零膨胀珀松分布和对数式珀松分布来模拟表达值。我们依据两种细胞类型所含细胞的比例进行随机扰动,分别得到最终的细胞数量。我们随机抽取5%的基因作为这种类型的DE基因。(ii) 基因在两个细胞类型下都是表达的,只是表达值高低不同。这种情形下,我们会随机模拟基因在两种细胞类型下表达值的倍数变化(Fold change)。第二种是两种不同实验条件下同一种细胞类型的差异表达基因。
第二种是同一种细胞类型在不同实验条件下之间的差异表达基因。这种情形,我们只需要根据多项分布生成每个细胞类型下的基因数量。每个细胞类型所含细胞的比例是基于整体中每个细胞类型的比例模拟而成。然后对每个细胞类型,分别利用每个细胞类型表达值分布的参数来估计该类型下基因的表达值。最后我们会分别得到一个细胞特异的基因表达矩阵。注意,在这种情形下考虑到建模的难度,我们不会专门预测DE基因。相反,我们会直接根据构造的细胞特异的基因表达矩阵预测DE基因。
在这一系列模拟的基因表达矩阵上,我们利用现有的主流算法预测DE基因。我们采用功效(Power)也叫真阳性率(True positive rate,TPR),和错误发现率(False discovery rate,FDR)来衡量预测的准确性。我们会尝试一系列不同的细胞数量,并从中发现能够确保FDR和TPR前提下,尽可能少的细胞数量,作为最终我们做实验用到的细胞数量。
输出结果:所需的最少细胞数量。
算法流程:
https://blog.sciencenet.cn/blog-3447504-1250216.html
上一篇:powsimR:RNA-Seq以及scRNA-Seq功效分析工具
下一篇:一张表弄懂细胞可塑性、细胞分化轨迹、细胞谱系和RNA速率