minimap2 -c ref.fa query.fa > alignment.c.paf
结果解释
.paf结果至少包括12列,加其他字段
| Col | Type | Description |
| 1 | string | Query序列ID |
| 2 | int | Query序列长度 |
| 3 | int | Query比对开始位置(*based) |
| 4 | int | Query比对结束位置(*based) |
| 5 | char | 如果query/target是正链关系,'+'表示;负链关系,'-'表示 |
| 6 | string | Target序列ID |
| 7 | int | Target序列长度 |
| 8 | int | Target比对开始位置 |
| 9 | int | Target结束开始位置 |
| 10 | int | 比对上的碱基数 (matching bases) |
| 11 | int | 对齐区域长度(包括gaps) |
| 12 | int | 比对质量(0-255) |
tags(the SAM-like typed key-value format)
| Tag | Type | Description |
| tp | A | aln类型:P/primary, S/secondary and I,i/inversion |
| cm | i | Number of minimizers on the chain |
| s1 | i | Chaining score |
| s2 | i | Chaining score of the best secondary chain |
| NM | i | Total number of mismatches and gaps in the alignment |
| MD | Z | To generate the ref sequence in the alignment |
| AS | i | DP alignment score |
| SA | Z | List of other supplementary alignments |
| ms | i | DP score of the max scoring segment in the alignment |
| nn | i | Number of ambiguous bases in the alignment |
| ts | A | Transcript strand (splice mode only) |
| cg | Z | CIGAR string (only in PAF) |
| cs | Z | Difference string |
| dv | f | Approximate per-base sequence divergence |
| de | f | Gap-compressed per-base sequence divergence |
| rl | i | Length of query regions harboring repetitive seeds |
2. SAM格式
命令
minimap2 -a ref.fa query.fa > alignment.a.sam
结果解释
SAM文件由两部分组成,头部区和主体区,都以tab分列。

1. 头部区:以’@'开始,体现了比对的一些总体信息。比如比对的SAM格式版本,比对的参考序列,比对使用的软件等。
2. 主体区:比对结果,每一个比对结果是一行,有11个主列和一个可选列。
主体区部分:
| 列 | 关键字 | 描述 |
| 1 | QNAME | Query序列ID |
| 2 | FLAG | Bwise FLAG(表示比对类型:paring,strand,mate strand等),如:0,99,256,2048等 |
| 3 | RENAME | Target序列ID |
| 4 | POS | 比对到参考序列上的位置,从1开始计数;未比对上为0 |
| 5 | MAPQ | 比对的质量分数(越高说明比对到参考序列的上的位置越准确)。如果是255,说明该比对值无效。 |
| 6 | CIGAR | 简要比对信息表达式 |
| 7 | MRNM | 下一片段比对上的参考序列编号 |
| 8 | MPOS | 下一片段比对上的位置,如果不可用,此处为0 |
| 9 | ISIZE | 插入片段长度 |
| 10 | SEQ | 和参考序列在同一个链上比对的序列(若比对结果在负义链上,则序列是其反向重复序列,反向互补序列) |
| 11 | QUAL | 比对序列的质量(ASCII-33=Phred base quality)reads碱基质量值 |
| 12 | Optional Fields | 可选的列以TAG:TYPE:VALUE的形式提供额外的信息 |
备注
1. FLAG释义表如下
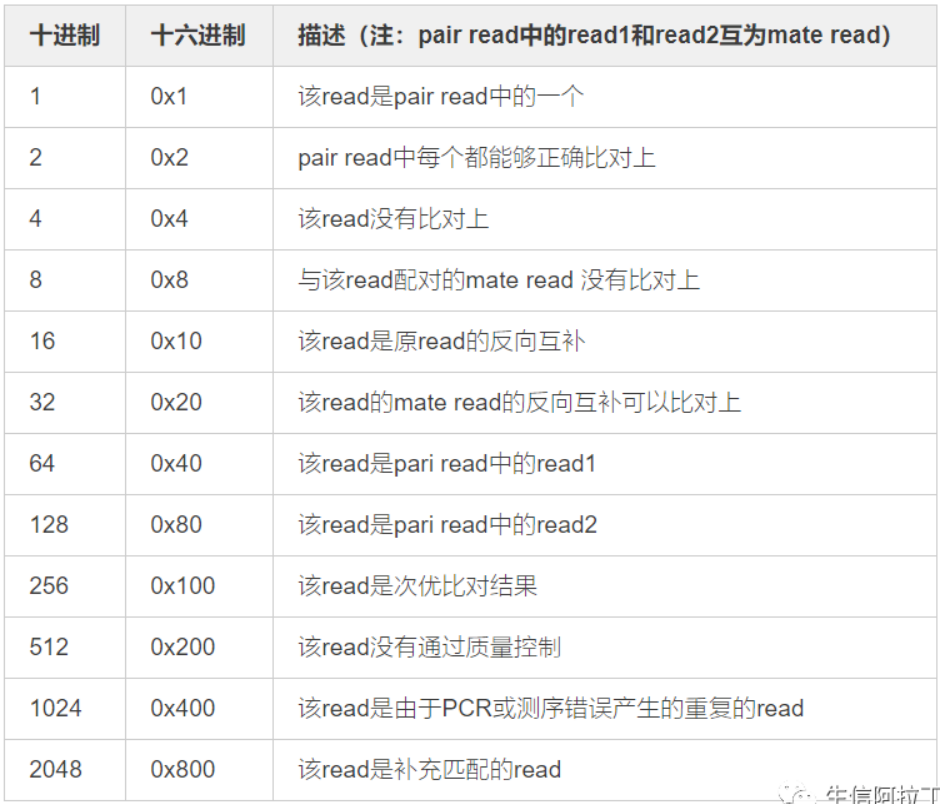
2. CIGAR string,简要比对信息表达式,示例如下图
