博文
代谢学人--Nature Cell Biology:铁死亡的导火索
||
代谢学人
Nature Cell Biology:铁死亡的导火索
撰文 | 徐梓禾 于柳 陈俊桐 徐鑫铭 许赛男 邱瑾
编辑 | 孟美瑶
校对 | 陈俊桐

一转眼
春天也悄然而至啦!柳树抽出嫩芽就像心中的希望也在渐渐破土而出了呢

春天应该是什么样子的?
是可以三三两两地在丽水河畔
静看樱花飘舞
亦或许独卧寝塌下
沐春风的惬意

百种场景
皆逃不出一份天真的勃勃生机
最美是云烟
看不够的是人间!
这份远道而来的蓬勃春意怎可浪费?
春风十里中
不吸收一点精神食粮怎么行呢?

你的春日好运正在派送
请保持心情舒畅哦~
让我们重拾状态
学习不停!
以饱满的热情
一起来欣赏本周的精读吧~
背景介绍
现今,随着工业化和城镇化的不断加快,以及慢性感染和不健康生活方式等危险因素的累积,全球癌症发病率正在逐步增加。癌症治疗也成为众人关注的问题。铁死亡是一种区别于细胞凋亡、细胞坏死、细胞自噬的新型的细胞程序性死亡方式。它是由铁依赖性脂质过氧化物的积累所导致的,并伴随着活性氧(ROS)升高的非凋亡的细胞死亡形式。ROS升高时超氧阴离子可以攻击脂质,造成脂质过氧化,使得生物膜成分&结构改变,造成脂膜损伤;ROS也可以氧化半胱氨酸,可导致蛋白失活,抑制酶活性;同时,ROS也可氧化嘌呤嘧啶,造成DNA氧化损伤,致使DNA链断裂,因此铁死亡过程通过大量产生的ROS损伤生物大分子,导致细胞死亡。
铁死亡在T细胞、B细胞及单核巨噬细胞等免疫细胞中发挥调节作用,研究发现,铁死亡作为抗肿瘤的一个新机制,可以被应用于肿瘤的免疫治疗,因而受到关注。铁死亡在T细胞介导的免疫治疗中的作用体现在CD8+T细胞分泌的干扰素γ(IFNγ)会下调肿瘤细胞中SLC3A2 和SLC7A11的含量,从而降低肿瘤细胞对胱氨酸的摄取,抑制了GPX4的信号通路,最终导致肿瘤细胞的铁死亡。同时,不同的B细胞亚群和巨噬细胞亚群对铁死亡敏感性有所差异,使GPX4在不同细胞亚群中抑制铁死亡的能力不同,最终影响B细胞和巨噬细胞的免疫反应。有研究表明,通过免疫疗法激活CD8+T细胞可以促进癌细胞中铁死亡特异性脂质过氧化物的积累,有助于免疫疗法的抗肿瘤效果。此外,电离辐射可诱导癌细胞内ROS升高和酰基辅酶A合成酶长链家族成员4(ACSL4)的表达,促进癌细胞发生铁死亡。
ACSL4是脂质过氧化所需的一种脂质代谢酶。在真核细胞质基质中,ACSL4可激活PUFAs形成长链酰基CoA,进而由溶血磷脂酰胆碱酰基转移酶3(LPCAT3)将长链酰基CoA插入质膜的磷脂中 。ACSL4可介导脂肪酸氧化的第一步--脂肪酸的活化,激活多不饱和脂肪酸(PUFA),尤其是花生四烯酸(C20:4)和肾上腺素酸(C22:4)生成长链脂酰基CoA。PUFA被活化后,能够作为高能化合物继续参与接下来的氧化过程,生成很多脂质过氧化中间产物,并通过LPCAT3的转运在生物膜中逐渐积累。改变了生物膜原有的脂质成分比例,造成生物膜结构的改变,使得细胞更易发生铁死亡。有研究表明,含有花生四烯酸(C20:4)和肾上腺素酸(C22:4)的磷脂可能是导致铁死亡的主要脂质过氧化底物。但是,对于这类引发铁死亡的脂质过氧化传感器和增强过程的报道则非常少。
蛋白激酶C(PKC)家族是在大多数细胞类型中发现的一组丝氨酸/苏氨酸激酶,其活性对多种信号转导途径有很强的影响,调节多种细胞的代谢、生长、增殖和分化。有研究表明,PKC与氧化应激之间存在着密切的联系。PKC作为蛋白激酶,含有多种亚型,包括α、βI、βⅡ和 γ亚类等。在本期介绍的研究中,作者发现PKCβII(PKC的亚型之一)是脂质过氧化的重要传感器,PKCβII的激活通过磷酸化和激活ACSL4放大了脂质过氧化。
Nature Cell Biology近期研究《PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis》表明,PKCβII激活是铁死亡的关键。PKCβII能够直接磷酸化ACSL4Thr328位点,而脂质过氧化–PKCβII–ACSL4正反馈机制可以增强脂质过氧化水平来诱导铁死亡。研究还证实,PKCβII–ACSL4机制通过调控铁死亡影响癌症免疫治疗的疗效,为铁死亡相关的癌症治疗提供了潜在的靶点和策略。

敲黑板啦!
1. PKCβII增强癌细胞对铁死亡的敏感性
2. PKCβII在铁死亡过程中被脂质过氧化诱导活化,是脂质过氧化的传感器
3.PKCβII直接磷酸化ACSL4 Thr328位点,促进铁死亡
4. ACSL4 Thr328磷酸化能激活ACSL4,促进含PUFA脂质的生物合成,积累脂质过氧化
5.PKCβII诱导的铁死亡提高了肿瘤免疫治疗的效果
研究结果
1.PKCβII是诱导铁死亡的关键因子
人们对调控铁死亡的分子和代谢基础知之甚少。为了确认调控铁死亡敏感性的因素,作者在乳腺癌细胞系MDA-MB-231中进行了规律间隔成簇短回文重复序列(CRISPR)-CRISPR相关蛋白9(Cas9)的筛选。在该模型中,作者通过铁死亡诱导剂erastin来抑制system-xc亚单位SLC7A11来诱导铁死亡(图1a(左)。使用铁死亡诱导剂诱导后,已知的铁死亡调控因子(包括ACSL4、GPX4、Keap1和PCBP1)均能被检测到,保证了筛查铁死亡敏感性的准确性(图1a(右)和附图1)。作者发现,诱导铁死亡后,细胞中PKCβ高表达(图1a(右),红色突出显示)。
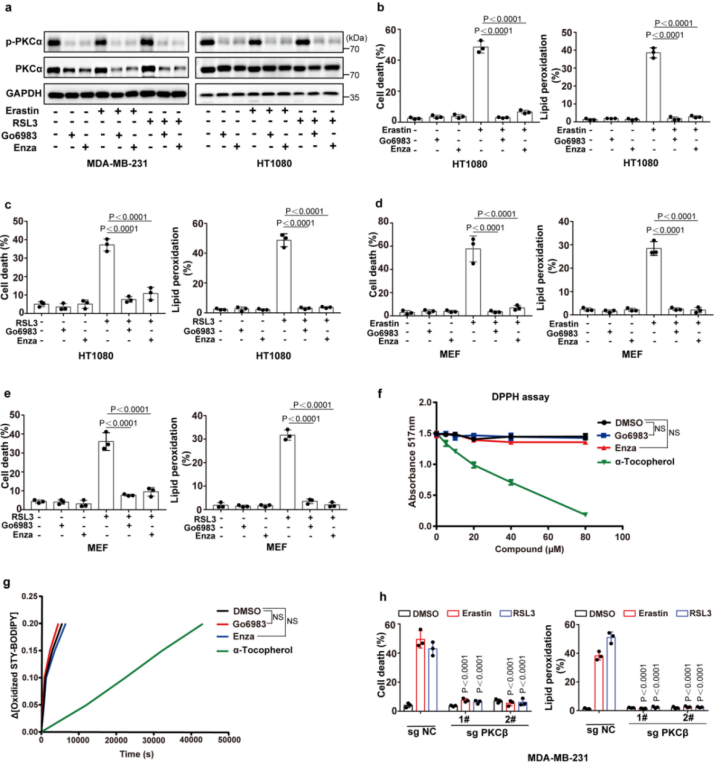
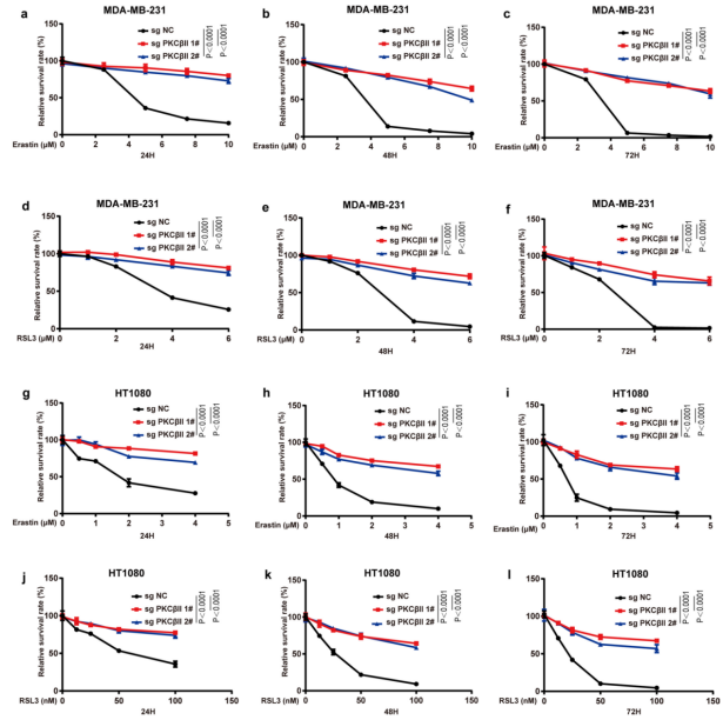
为了证实CRISPR–Cas9筛选的结果,作者使用了一个包含644种化合物的激酶抑制剂库进一步筛选(图1b(左))。作者发现有14种抑制剂可以抑制一半以上由erastin诱导的铁死亡,大多数为JAK、TRK、SRC和PKC抑制剂(图1b(右)和附图2)。其中,PKC抑制剂对铁死亡的抑制作用最强,这与先前CRISPR–Cas9筛选的结果一致。这两个独立的筛选结果说明PKCβ极有可能是诱发铁死亡的关键因素。紧接着,作者使用两种PKC抑制剂来进一步地证实,并发现Go6983和enzastaurin显著地抑制了erastin或RSL3处理诱发的脂质过氧化和铁死亡(图1c、d和附图1a-e)。此外,作者又进行了2,2,-二苯基-1-吡啶肼和抗氧化荧光检测试验,来检验Go6983和enzastaurin是否作为单纯的自由基捕获抗氧化剂来抑制细胞铁死亡。结果显示,Go6983和enzastaurin均未降低2,2-二苯基-1-吡啶肼溶液在517 nm处的吸光度,同样也未延缓氧化STY-BODIPY的生成,表明Go6983和enzastaurin并不是自由基捕获抗氧化剂(小编注:铁死亡伴随着脂质过氧化,而脂质过氧化发生的很重要的原因是自由基的产生,即ROS。这里想要说明这两种抑制剂是通过抑制了PKCβII的活性,来抑制了铁死亡,证明了PKCβII和铁死亡是有关系的。这个实验就是排除了干扰,排除了单纯清除自由基来抑制铁死亡的事件。)(附图1f,g)。由于Go6983和enzastaurin是泛PKC抑制剂,其靶点包括PKCα、PKCβ、PKCγ、PKCδ和PKCζ。为了确定PKCβ亚型是否调控铁死亡,作者检测了各个PKC亚型敲除后对erastin诱导的铁死亡的潜在抑制作用。结果显示,敲除PKCβ基因可以显著抑制erastin和RSL3诱导的铁死亡和脂质过氧化,而敲除其他PKC亚型基因却没有这种抑制效果(图1e、f和附图1h、附图2)。总之,这些数据表明,PKCβ对铁死亡过程非常重要。由于PKCβ有两个转录本,即PKCβI和PKCβII。为了检测哪种PKCβ的转录本调控了铁死亡,作者通过在sgRNA识别位点引入沉默突变,构建了具有sgRNA抗性的PKCβI或PKCβII来恢复PKCβ敲除的MDA-MB-231细胞PKCβI/PKCβII的表达,结果显示恢复PKCβII几乎完全逆转了PKCβ敲除对erastin和RSL3诱导的脂质过氧化和铁死亡的抑制作用(图1g)。随后,用erastin或RSL3,结合差异调控细胞铁死亡抑制剂来处理PKCβII回复后的细胞。正如预期的那样,铁死亡抑制剂ferrostatin-1(Fer-1;脂质过氧化清除剂)、铁螯合剂去铁胺(DFO)或抗氧化剂N-乙酰半胱氨酸可以完全回复由erastin和RSL3诱导的脂质过氧化和铁死亡,但细胞凋亡和坏死性凋亡抑制剂不能产生此作用(图1h-k和2a-d),说明PKCβII可以通过调控铁死亡来诱导细胞死亡。
为了探寻PKCβII在调控铁死亡中的普遍作用,作者在一系列癌细胞系中进行PKCβII基础表达与铁死亡敏感性的相关性分析。分析结果显示,相比于PKCβII低表达的细胞,PKCβII高表达的细胞对铁死亡的诱导物更加敏感(图2e-g),这意味着PKCβII是监测铁死亡的有效生物标志物。为了进一步证实PKCβII对癌细胞铁死亡敏感性的影响,作者在HS578T和MDA-MB-468细胞(富有PKCβII的癌细胞)中敲除了PKCβII。结果显示在HS578T和MDA-MB-468细胞中,PKCβII敲除显著抑制了由erastin和RSL3诱导的铁死亡和脂质过氧化(附图3a-l)。相反,作者在MCF-7和BT474细胞(PKCβII缺陷癌细胞)中过表达PKCβII,发现PKCβII的过表达显著增加了这些细胞中erastin和RSL3诱导的铁死亡和脂质过氧化(附图3m-x)。这些结果表明,PKCβII能够直接影响癌细胞对铁死亡的敏感性,是铁死亡的一个重要诱导物和生物标志物。
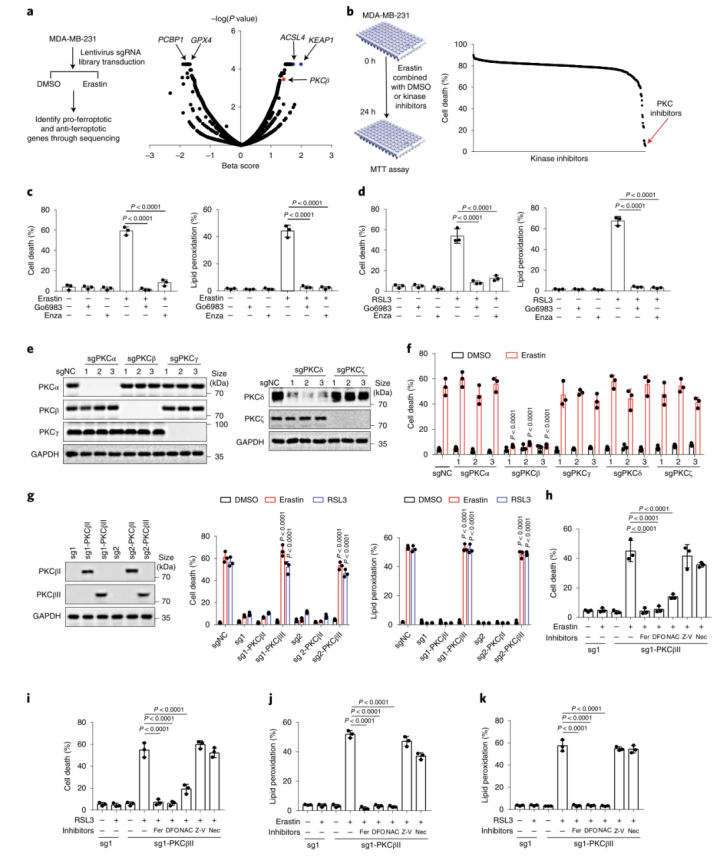
图1 PKCβII是铁死亡的关键因素
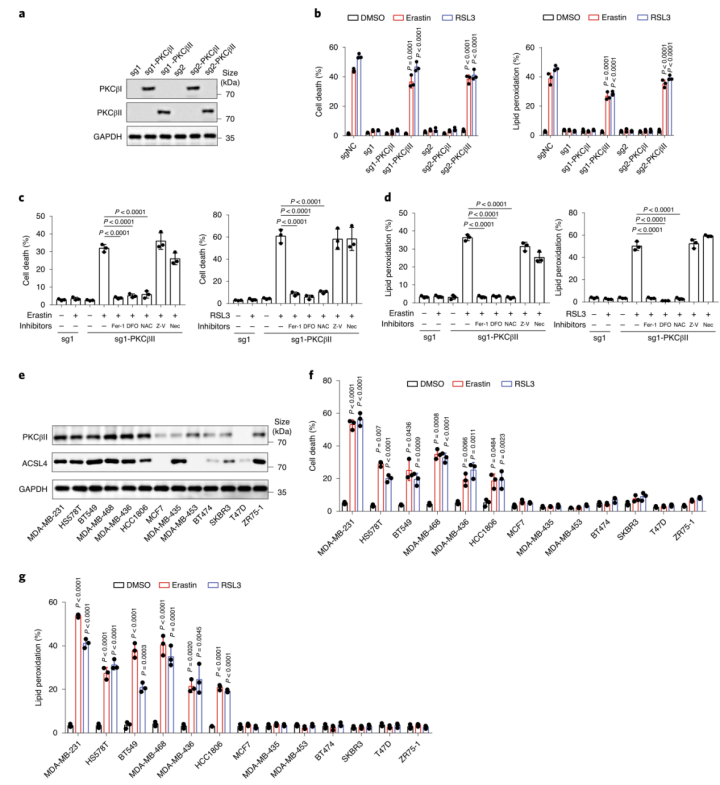
图2 PKCβII是铁死亡的生物标志物
附图1
附图2
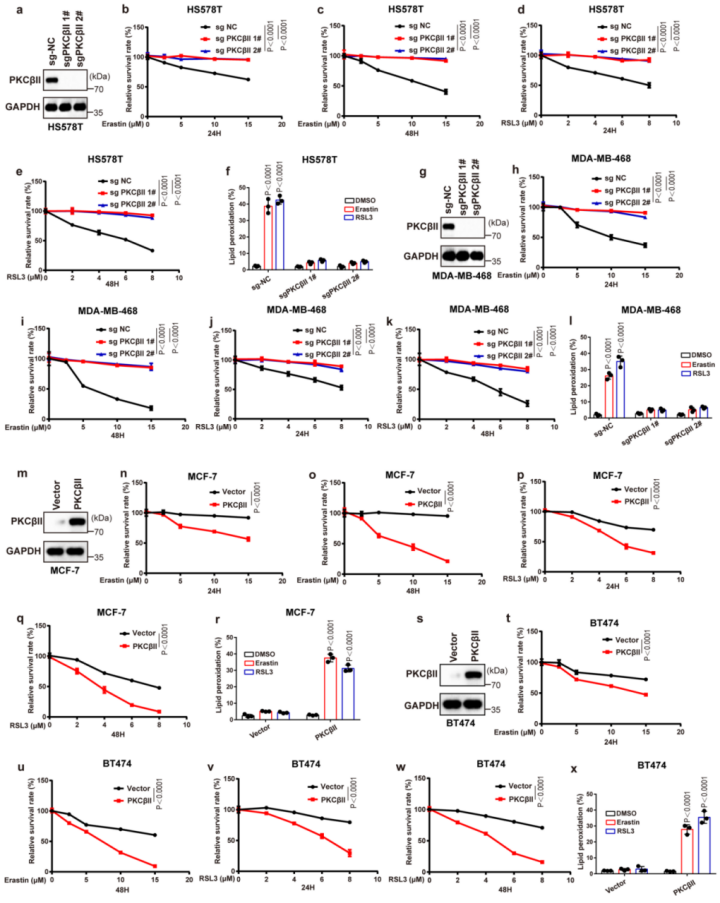
附图3
拓展阅读
CRISPR/Cas9文库筛选技术及激酶抑制剂筛选技术
CRISPR /Cas9文库筛选技术,是利用 CRISPR/Cas9技术建立哺乳动物全基因组突变库或者与某类功能相关的基因突变体库,通过功能性筛选和富集以及随后的 PCR 扩增和深度测序分析,发掘与筛选表型相关的基因的研究技术。 CRISPR /Cas9技术通过将重复间隔阵列整合到 CRISPR 中来识别和记录外源 DNA 或 RNA 片段。进而,CRISPR 前体被转录并加工成成熟的 CRISPR衍生 RNA (crRNA)。在 crRNA 间隔区和互补的侵入性核苷酸序列杂交后,cas 核酸内切酶造成了与原型间隔区相邻基序(PAM)相邻的双链断裂(DSB)。再通过非同源末端连接(NHEJ)和同源定向修复(HDR)修复 DSB,使用同源修复模板导致末端连接、碱基插入、缺失或定向突变。在癌症治疗中, CRISPR/Cas9筛选已被用于识别不同细胞环境中肿瘤的新靶标。由于CRISPR/Cas9的大规模筛选并不局限于细胞系统,研究人员已成功在体内进行CRISPR/Cas9文库筛选。CRISPR/Cas9文库筛选技术加速新药靶点和生物标志物的识别,促进了精准癌症治疗的发现和开发。本篇文章中,作者就利用了CRISPR/Cas9文库筛选技术,在诱导铁死亡的条件下,找到了一系列铁死亡调控因子,其中包括了PKCβ。激酶抑制剂可阻断某些与疾病相关的酶的活性,如癌症和炎症性疾病。激酶抑制剂库包含激酶抑制剂和调节因子,主要靶向对蛋白激酶 (VEGFR、EGFR、BTK、CDK、Ak t等)、脂质激酶 (PI3K、PI4K、SK 等) 和碳水化合物激酶 (己糖激酶) ,是激酶药物开发及相关研究的有用工具,本文利用激酶抑制剂库筛选来进一步确定了PKCβII和铁死亡之间的关系。
参考文献:[1]Xing, Hui,Acta pharmacologicaSinica vol. 41,5 (2020): 583-587.[2]ZhanTZ, Rindtorff N, Betge J, Semin Cancer Biol. 2019;55:106–19.
经典PKC家族(Classical PKC family)
经典蛋白激酶C(PKC)家族是一类丝氨酸和苏氨酸的特异性蛋白激酶,在人类中以PKCα、PKCβ、PKCγ形式存在,通过磷酸化多种蛋白质靶点参与细胞增殖、分化、凋亡和血管生成的信号转导。有研究发现,PKCβ与致癌性和维持恶性表型方面有关。PKCβ基因可以编码两种不同的蛋白质,分别为PKCβI和PKCβII。它们的区别在于最后的50个氨基酸,从而实现了相互对立的细胞功能:PKCβI与细胞分化有关,而PKCβII参与细胞增殖的过程。本研究中介绍的是PKCβII,它是血管内皮生长因子诱导的内皮细胞增殖的主要媒介,而血管内皮生长因子是乳腺癌中肿瘤血管生成和生长的一个刺激因素。靶向PKCβ抑制剂的药物能够为乳腺癌的治疗提供了方向和思路。本研究中探索了PKCβII对ACSL4的激活作用,从分子机制上解释了PKCβ对癌细胞的影响途径,从而为研制PKCβ抑制剂来针对癌症治疗提供进一步的证据。
参考文献:[1]Koivunen J, Aaltonen V, Peltonen J. Cancer Lett. 2006;235(1):1-10[2]Sledge GW Jr, Gökmen-Polar Y. Semin Oncol.2006;33(3Suppl9):S15-S18.
2. PKCβII激活对铁死亡至关重要
PKC广泛分布于多种组织、器官和细胞中。静止细胞中PKC主要存在于胞浆中,呈非活性构象。当细胞受到刺激(氧化应激)后,PKC以Ca2+依赖的形式从胞浆中移位到细胞膜上(即转位),成为膜结合的酶,进而能磷酸化并激活细胞质中的酶,参与生化反应的调控。接下来,作者试图评估PKCβII是否在铁死亡过程中被激活。在MDA-MB-231和HT1080细胞中,PKCβII磷酸化增加结合膜定位结果表明erastin和RSL3可以浓度和时间依赖的诱导PKCβII激活(图3a-f和附图4a,b)。相比之下,erastin处理没有增加其他PKC亚型的激活(扩展数据图4c)。为了进一步的分析PKCβII对细胞铁死亡的潜在作用,作者将含有野生型PKCβII(PKCβII-WT)或其激酶失活型突变体(PKCβII-KD;PKCβIIK371R)的质粒转染到PKCβ敲除的MDA-MB-231和HT1080细胞中。结果显示:与稳定转染PKCβII-WT的细胞相比,稳定转染PKCβII-KD的细胞中由erastin和RSL3诱导的脂质过氧化和铁死亡程度显著降低(图3g、h和附图4d),这表明PKCβII依赖激酶活性来促进铁死亡。此外,在MDA-MB-231和HT1080细胞中,脂质过氧化清除剂Fer-1和liproxstatin-1(Lipro-1)能够完全抑制PKCβII的激活(图3i-l和附图4e-h)。这些结果表明,脂质过氧化在铁死亡过程中能够诱导PKCβII活化,并且意味着PKCβII可能是与铁死亡相关的脂质过氧化传感器。总之, PKCβII的激活对铁死亡的发生至关重要。
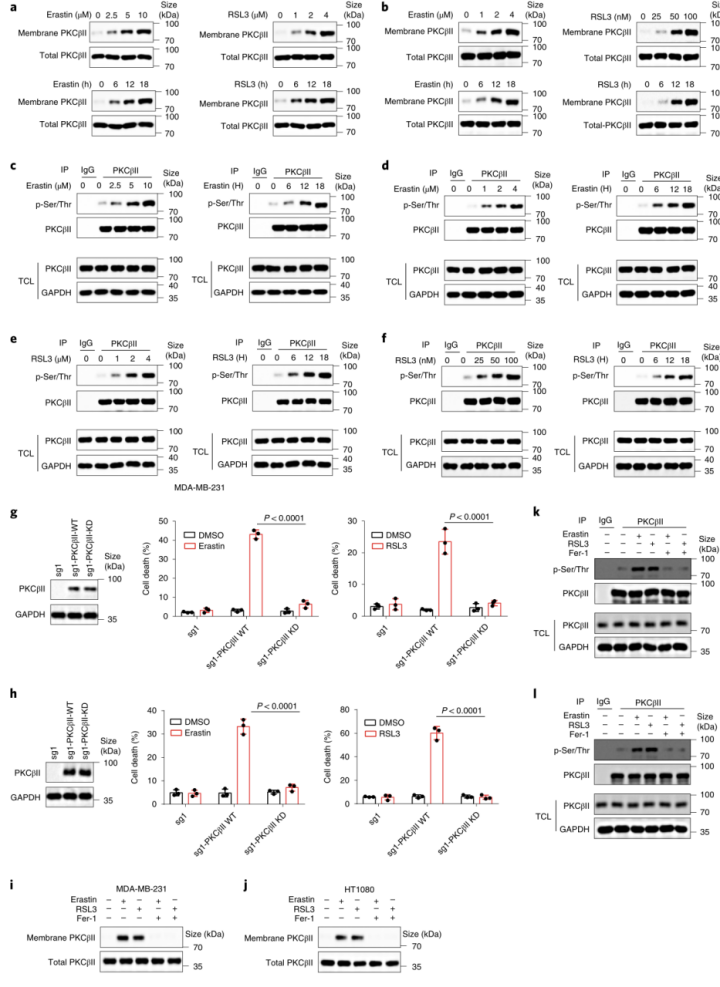
图3 PKCβII激活对铁死亡至关重要
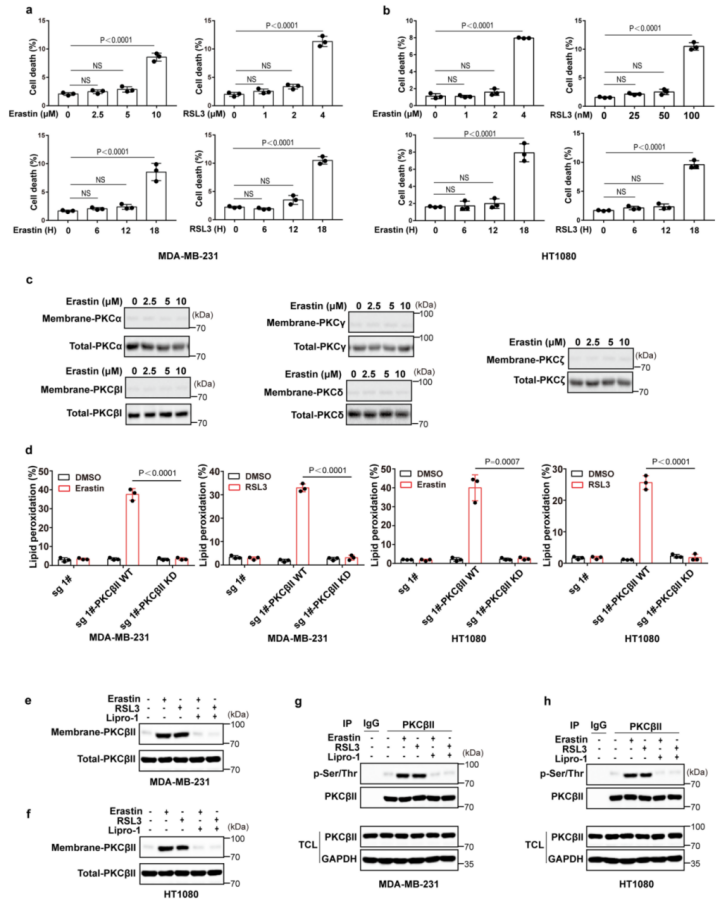
附图4
3.PKCβII和ACSL4相互作用并直接磷酸化ACSL4
上述数据表明,抑制PKC显著地阻碍了由铁死亡诱导剂处理导致的脂质过氧化产物的增加。作者假设PKCβII可能通过影响脂质过氧化产物的产生或清除来调控铁死亡。结果表明,PKCβII敲除或PKC抑制剂的添加都不影响GPX4(小编注:GPX4蛋白对脂质过氧化物有高度偏好性,利用其催化活性可以清除膜脂上过氧化氢产物,维持膜脂质双分子层稳态,从而保护细胞免受膜脂质过氧化和细胞死亡)的蛋白质水平和细胞内的二价铁水平(附图5a-c)。因此,作者进一步研究了PKCβII是否通过影响ACSL4的活性来调控铁死亡。免疫沉淀结果显示,在MDA-MB-231和HT1080细胞中,erastin或RSL3(铁死亡诱导剂)处理能显著地增强ACSL4和PKCβII(小编注:体外研究表明,ACSL4可以作为PKC的底物,PKC激酶可以控制其磷酸化进而调控其活性。[1]但是体内是否会发生ACSL4作为PKCβII的底物被磷酸化的情况是在本文被证明的。参考文献:[1]Smith ME,et al. Biochem Biophys Res Commun. 2013)之间的相互作用,该相互作用呈现出浓度依赖性;然而,当添加Go6983或enzastaurin(PKC抑制剂)时,这种效应显著降低,表明PKCβII的激酶活性有利于PKCβII–ACSL4复合物的形成(图4a,b)。于是,作者推测PKCβII可能通过磷酸化和激活ACSL4来促进铁死亡。为了验证该推论,检测发现erastin或RSL3(铁死亡诱导剂)处理后ACSL4的总磷酸化水平显著增强,并且这增强的磷酸化水平还可被Go6983和enzastaurin(PKC抑制剂)抑制(图4c,d)。随后,作者结合GPS(Group-basedPrediction System,磷酸化位点预测系统)和SCANSITE(蛋白质底物位点检索与预测工具)程序去预测ACSL4的哪些残基可能被PKCβII磷酸化(附图5d)。通过建立敲入ACSL4基因的细胞模型,作者进一步检测了ACSL4每个磷酸化位点对erstin诱导的铁死亡的影响:即将预测出的ACSL4潜在的磷酸化位点突变为不可磷酸化的Ala(磷酸化失活突变,Ser/Thr突变为Ala;附图5e),来进一步检测各个磷酸化位点对erastin诱导的铁死亡的影响。令人惊讶的是,与含有其他位点突变或野生型ACSL4的细胞相比,表达ACSL4-Thr328Ala突变体的细胞中erastin诱导的铁死亡程度显著降低(图4e)。这些结果表明,PKCβII可能通过磷酸化ACSL4的Thr328位点来促进铁死亡。为了进一步证实该结论,作者进行体外激酶实验,来分析PKCβII是否直接磷酸化ACSL4的Thr328位点。结果显示:在存在重组活性PKCβII激酶的情况下,野生型ACSL4(ACSL4-WT)蛋白的Ser/Thr磷酸化水平显著增加,而ACSL4-Thr328Ala蛋白中磷酸化作用被阻断(图4f,g)。接着,作者进一步研究了内源性Thr328磷酸化作用,结果发现:通过使用磷酸化Thr328-ACSL4特异性抗体p-ACSL4(Thr328)检测出PKCβII能够使野生型ACSL4磷酸化,但并不能使ACSL4-Thr328Ala突变体磷酸化(图4h,i)。另外,PKCβII的缺乏或失活抑制了铁死亡诱导物对ACSL4Thr328的磷酸化作用,这表明铁死亡诱导物介导的ACSL4Thr328处的磷酸化是依赖PKCβII而发生的(图4j,k)。此外,作者还发现,使用Fer-1或Lipro-1(脂质过氧化清除剂,即铁死亡抑制剂)能显著抑制erastin和RSL3诱导的ACSL4磷酸化(附图5f-i)。这些结果表明,Fer-1和Lipro-1通过消除脂质过氧化物来阻断PKCβII–ACSL4通路。总之, PKCβII通过直接磷酸化ACSL4Thr328来促进铁死亡。
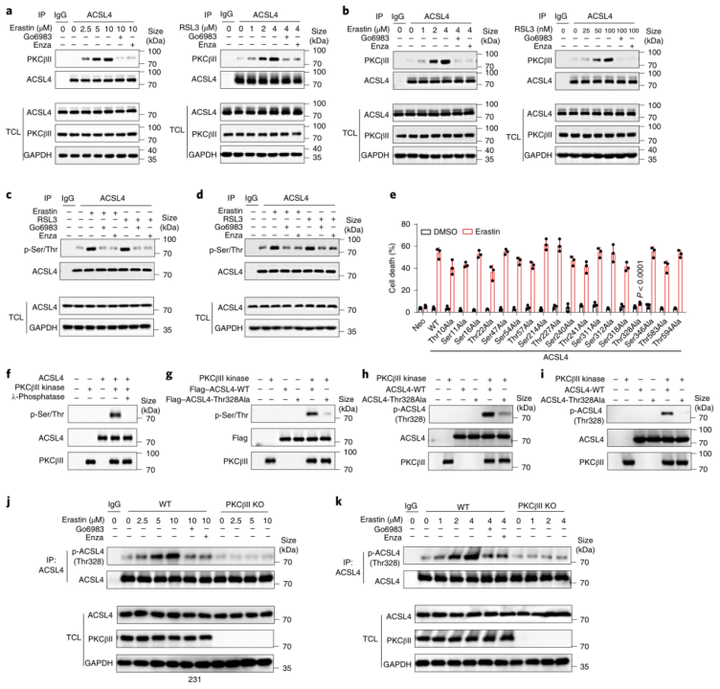
图4 PKCβII和ACSL4相互作用并直接磷酸化ACSL4
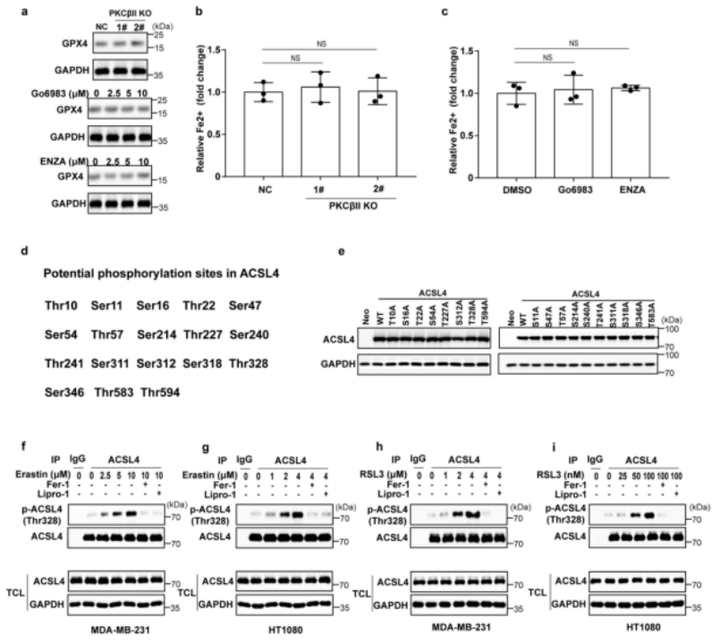
附图5
4.Thr328磷酸化对ACL4的激活至关重要
ACSL4 Thr328磷酸化在铁死亡中的生物学功能被揭示。研究表明,ACSL4的二聚化对于ACSL4酶的激活十分重要。作者发现,经erastin或RSL3(铁死亡诱导剂)处理后,PKCβII的敲除降低了MDA-MB-231细胞中的ACSL4二聚化作用(图5a(左),b)。与此同时,与ACSL4-WT相比,经erastin和RSL3处理后,ACSL4-Thr328Ala突变体的ACSL4二聚化水平降低(图5a(右),c)。这表明PKCβII介导的ACSL4Thr328磷酸化影响了ACSL4的二聚化进而影响了其催化活性。此外,作者发现使用Fer-1或Lipro-1(铁死亡抑制剂)后,erastin和RSL3诱导的ACSL4二聚化被显著抑制(附图6a,b)。总之,PKCβII介导的ACSL4Thr328处的磷酸化促进了ACSL4的二聚化和激活。作者推测,PKCβII介导的ACSL4磷酸化可能激活ACSL4,促进含有PUFA的磷脂生成,从而导致铁死亡。为了评估这种可能性,作者对重新表达ACSL4-WT和ACSL4-Thr328Ala的细胞系进行erastin(铁死亡诱导剂)或enzastaurin(PKC抑制剂)处理,或者用两者同时处理细胞系,进行非靶向脂质组学分析。结果显示:在重新表达ACSL4-WT的细胞中,erastin处理大大增加了PUFA磷脂(磷脂酰乙醇胺(PE)18:0_20:4和PE18:0_22:4)的水平,而增加的PUFA磷脂在enzastaurin处理后显著减少(图5d,e和补充表3,4)。这表明PKCβII在铁死亡过程中诱导了含PUFA磷脂的生物合成。相反地,在重新表达ACSL4-Thr328Ala的细胞中,erastin处理后的细胞系中降低了含有PUFA的磷脂水平。这说明在铁死亡过程中,PKCβII介导的ACSL4 Thr328的磷酸化对于PUFA磷脂的生物合成很重要。此外,作者发现在重新表达ACSL4-WT的细胞和重新表达ACSL4-Thr328Ala的细胞之间,单不饱和脂肪酸和饱和脂肪酸脂质的基础水平没有差异,并且这些水平不受erastin处理的影响(附图6c和补充表3,4)。总之,PKCβII–ACSL4途径通过增加含PUFA的磷脂产量来促进铁死亡。进一步的实验证实,给重新表达ACSL4-WT的细胞补充花生四烯酸(C20:4)或肾上腺素酸(C22:4),能显著增加erastin和RSL3诱导的脂质过氧化和铁死亡,但重新表达ACSL4-Thr328Ala的细胞无法产生此效应(图5f)。所以,ACSL4Thr328的磷酸化对ACSL4介导的含有花生四烯酸(C20:4)和肾上腺素酸(C22:4)的PEs的产生起到了关键作用,而这些PEs能促进铁死亡。为了证实这些发现,作者将载体对照、ACSL4-WT、ACSL4-Thr328Ala或ACSL4-Thr328Asp(小编注:拟磷酸突变,即当丝氨酸突变为丙氨酸,氨基酸永远都不会被挂上磷酸基团,而假如突变为天冬氨酸,天冬氨酸自己带了个磷酸根,所以就模拟了氨基酸被磷酸化的状态,称为拟磷酸突变)的质粒稳定转染到ACSL4敲除的MDA-MB-231和HT1080细胞中(附图7a)。结果显示:与ACSL4-WT和ACSL4-Thr328Asp细胞相比,ACSL4-Thr328Ala细胞中erastin和RSL3诱导的脂质过氧化和铁死亡程度均显著降低(图5g、h和附图7b、c)。同时,PKCβII敲除细胞中ACSL4-Thr328Asp变体的稳定表达足以使细胞对铁死亡恢复敏感(附图8a-j)。由此可知,PKCβII介导的Thr328磷酸化对于ACSL4的激活和促进铁死亡功能很关键。此外,在缺血再灌注过程中,ACSL4Thr328磷酸化逐渐增加(附图8k-m)。由此分析,ACSL4Thr328磷酸化可能对鉴别不同病理模型的铁死亡非常有用(小编注:多数情况下,缺血后再灌注可使组织器官功能得到恢复、损伤的结构得到修复,使患者病情好转康复;但有时缺血后再灌注,不仅不能使组织器官功能恢复,反而加重其功能障碍和结构损伤。本研究中缺血再灌注后,发现ACSL4 Thr328磷酸化逐渐增加,加剧了体内细胞发生铁死亡这一过程。导致铁死亡发生的病理机制(信号通路)有很多,通过对缺血再灌注后不同信号指标的检测,可以鉴定当前研究的铁死亡的分属类型)总之,PKCβII介导的ACSL4Thr328磷酸化可以促进含PUFA脂质的生物合成,从而促进脂质过氧化的积累,导致铁死亡。研究表明,在铁死亡过程中脂质过氧化诱导PKCβII的活化。凭借PKCβII–ACSL4机制,活化的PKCβII通过磷酸化和激活ACSL4来进一步促进脂质过氧化产物的增加。PKCβII能识别到最初的脂质过氧化物,并激活增加和铁死亡相关联的脂质过氧化。脂质过氧化–PKCβII–ACSL4正反馈机制促进增加脂质过氧化,在铁死亡过程中起到了核心作用。
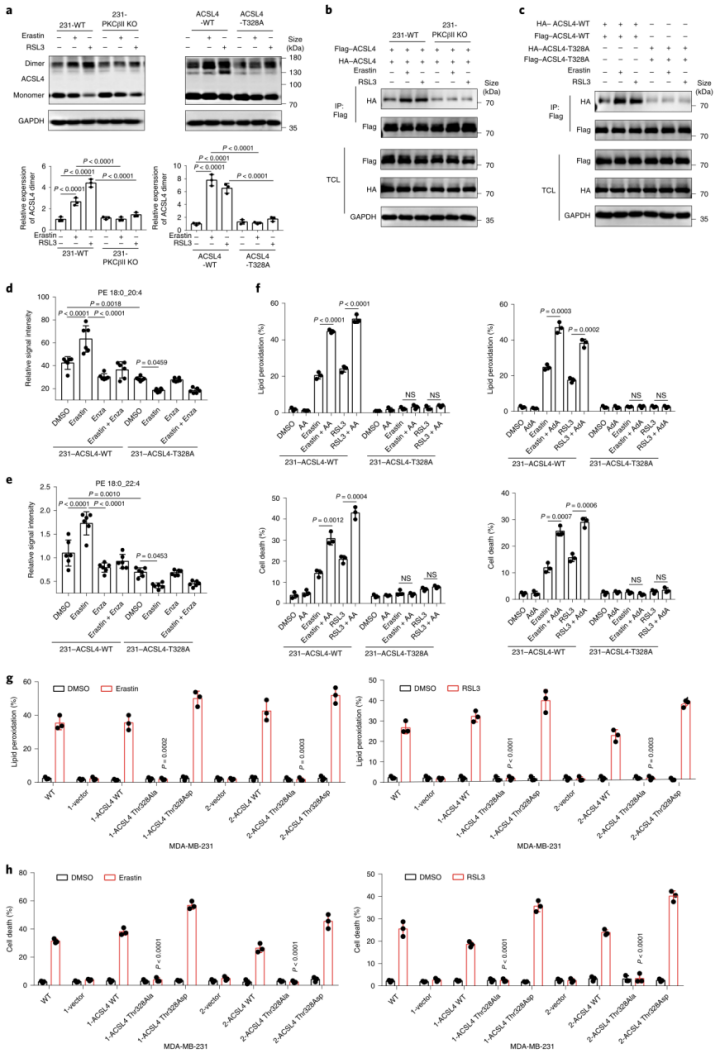
图5 Thr328磷酸化对ACSL4的激活和促铁死亡功能至关重要
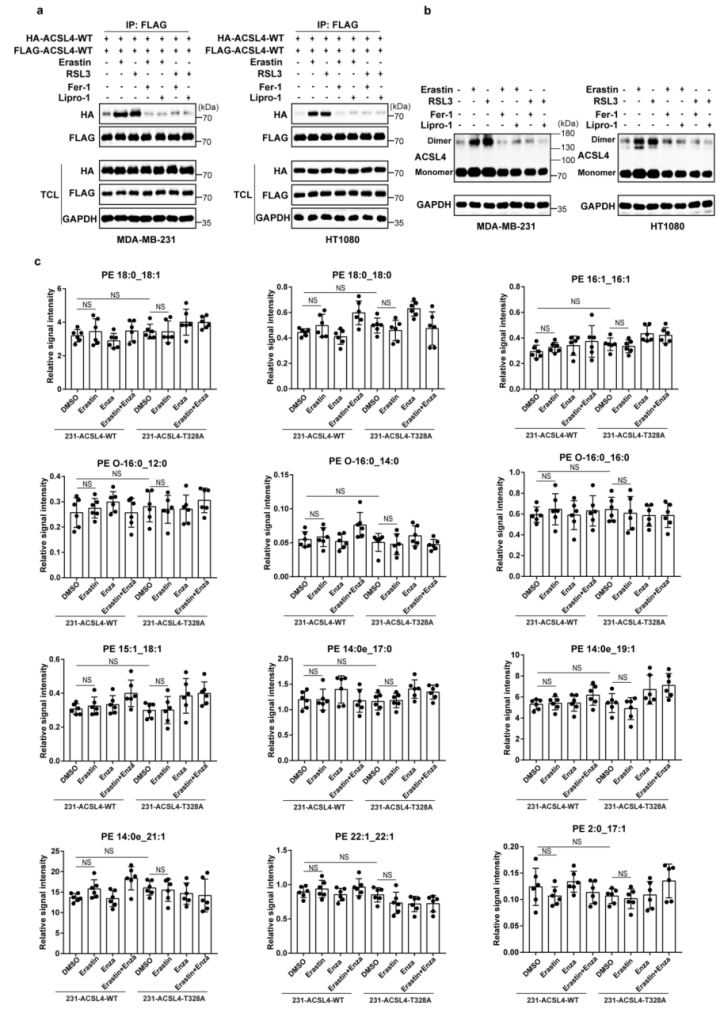
附图6
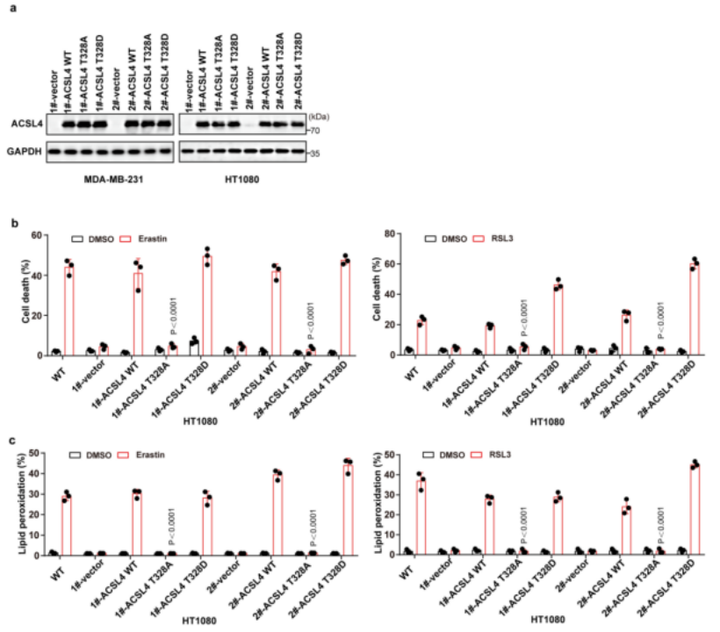
附图7
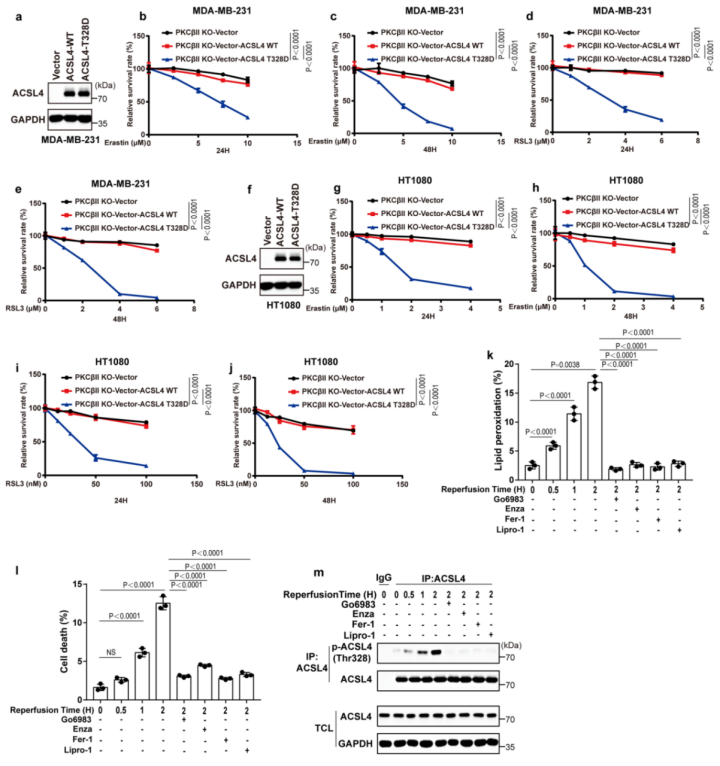
附图8
拓展阅读
PUFA与铁死亡敏感性的关系
最初研究认为含花生四烯酸(AA)或肾上腺酸的磷脂酰乙醇胺(PE)是铁死亡中脂质过氧化的主要底物,而近期数据则指向广泛的多不饱和脂肪酸(PUFAs),此处PUFA由于其较多的双键结构更易被超氧阴离子活化,更易作为脂质过氧化物来诱导铁死亡,其他脂质报道较少;PUFA是在FA产生后,由去饱和酶和延伸酶的协同作用来合成的。
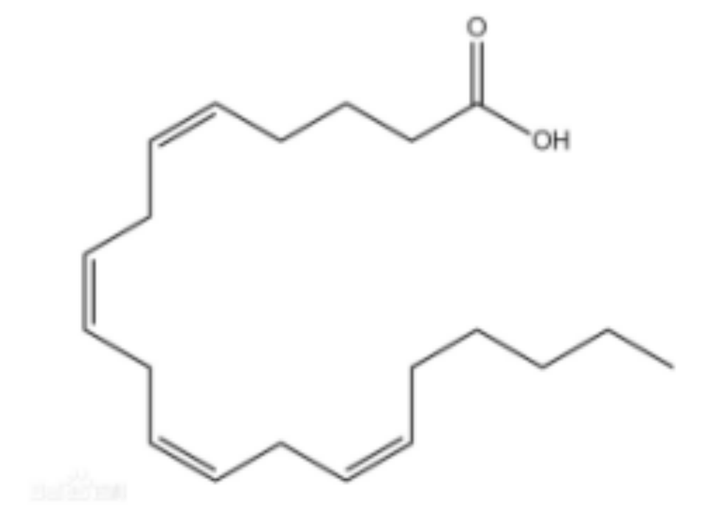
花生四烯酸

肾上腺酸
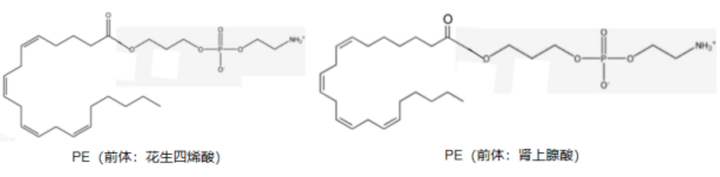
PE
哺乳动物细胞中的PUFAs主要为食源性必需脂肪酸(亚油酸和α-亚麻酸)经去饱和酶和延伸酶的协同作用转化而来。去饱和酶2敲低的肝癌细胞能够抵抗RSL3诱导的脂质过氧化,与PUFAs延伸有关的丙二酰辅酶A的合成途径对铁死亡起关键性的正向调控,此外,丙二酰辅酶A对脂肪酸β氧化的抑制作用也促进铁死亡的发生。由此可知,PUFA的合成或降解过程将最终影响细胞对铁死亡的敏感性。
研究表明,在铁死亡情况下,脂质过氧化影响的主要是酯化的PUFA,而非游离的PUFA。
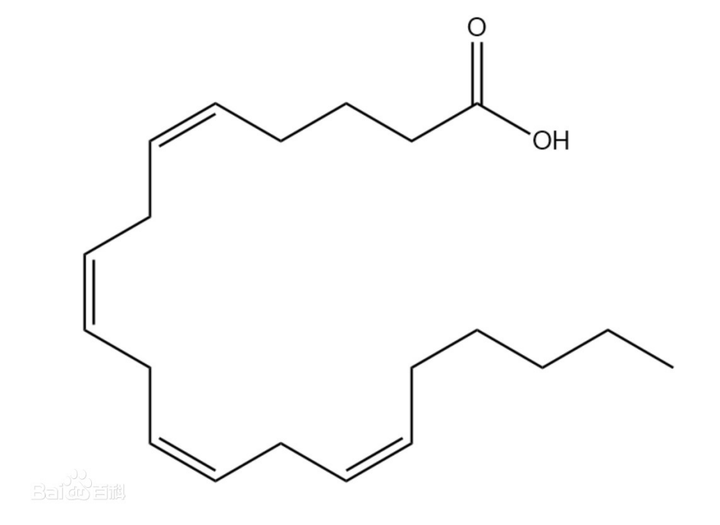
花生四烯酸(游离的/未活化)
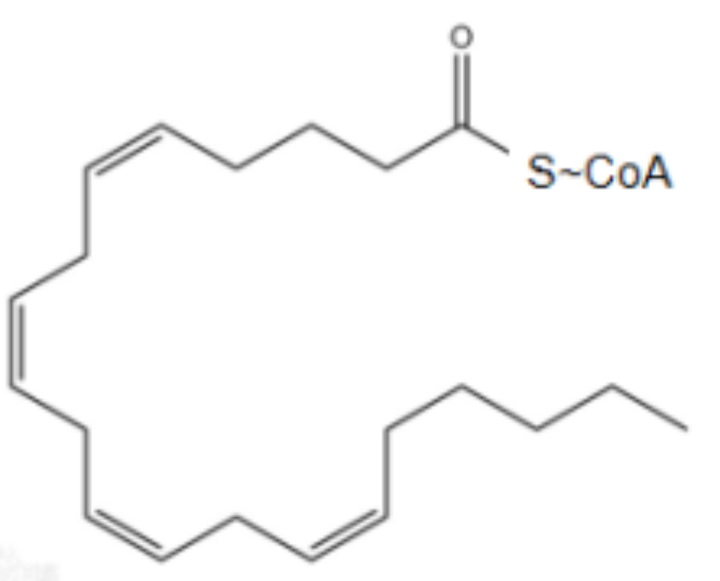
花生四烯酸酰基辅酶A(酯化)
游离PUFA是合成脂质信号传导介质的底物,但是必须要被酯化为膜磷脂(双分子层膜,磷脂酰乙醇胺)并进行氧化才能转化为有效信号,来介导铁死亡,形成正反馈。细胞除了依靠磷脂酶PLA2和溶血磷脂酰丝氨酸脂酶ABHD12水解磷脂上的PUFA来避免脂质过氧化,也可以利用外源性单不饱和脂肪酸(MUFA)取代磷脂PUFA。此外,MUFA生物合成需要硬脂酰CoA去饱和酶(SCD)将饱和脂肪酸转化为MUFAs。本文实验就可以利用SCD1过表达表现出对铁死亡的抵抗力,模拟MUFA治疗的效果。总之,磷脂中的PUFAs充足是铁死亡插手细胞命运的先决条件。
参考文献:
[1]Jiashuo Zheng, et al. Cell Metabolism. 2020.
Acsl4结构与活性调节
Acsl4是一个由多种分子间盐桥相互作用形成的同型二聚体,能够催化12~20个碳长度的脂肪酸、ATP、辅酶A,生成长链酰基辅酶A。Acsl4通常位于细胞质基质中,可作为PKA和PKC的底物被磷酸化激活,进而参与下游生化效应。Acsl4可同时被多种激酶磷酸化,且每一个的磷酸化可以是独立的,发生在不同的位点。
有研究表明,Acsl4的激活和二聚体无明显相关性,Acsl4二聚化可以发生在其被磷酸化作用之前。但Acsl4作为二聚体,其形成的蛋白空间结构能够帮助其更好的结合底物,从而有效地发挥催化作用。本文中作者发现使用Fer-1或Lipro-1(铁死亡抑制剂)后,erastin和RSL3诱导的ACSL4二聚化被显著抑制,说明ACSL4与铁死亡过程相关,ACSL4催化活性被抑制,铁死亡过程也相应地被抑制。
参考文献:
[1]Smith,M. E., et al. Biochem. Biophys. Res.Commun. 430, 272–277 (2013).
5.PKCβII–ACSL4通路增强免疫治疗的效果
肿瘤免疫治疗被认为是一种重要的体内铁死亡相关病理模型。作者获得了49例黑色素瘤患者的公开数据(GSE91061),这些患者在开始免疫治疗之前接受了肿瘤活检。总结数据来看,32.1%的“ACSL4高”组患者对nivolumab(纳武单抗OPDIVO,一种PD-1抑制剂,可以改善肿瘤的免疫抑制反应,从而达到治疗癌症的作用,而“ACSL4低”组中只有4.8%的患者对nivolumab敏感。这表明表达低ACSL4的肿瘤对免疫检查点抑制剂具有耐药性(补充表5)。紧接着,作者进一步研究了体内抗PD1治疗后ACSL4Thr328磷酸化对肿瘤生长的影响。全身性PD1抗体靶向治疗显著提高了脂质过氧化以及活化的细胞毒性CD4+T和CD8+T细胞水平。并且在重新表达ACSL4-WT的B16-ACSL4基因敲除肿瘤异种移植的免疫活性C57小鼠模型中,系统性PD1抗体靶向治疗抑制了肿瘤生长。但在重新表达ACSL4-Thr328Ala的小鼠模型中系统性PD1抗体靶向治疗不能够抑制肿瘤生长(图6a-e和附图9a)。这表明ACSL4的Thr328磷酸化失活突变能通过抑制铁死亡来削弱免疫治疗的效果。另外,作者在4T1(小鼠乳腺癌细胞)野生型(4T1-WT)和4T1-PKCβII基因敲除型肿瘤细胞(图6f-h和附图9b,c)之间比较抗PD1的抗肿瘤效果时,获得了类似的结果。总之,PKCβII–ACSL4通路的减弱抑制铁死亡,削弱了免疫治疗的效果。(小编注:研究发现,过表达ACSL4会加重缺血性脑损伤;ACSL4过表达也可通过增强脂质过氧化促进了神经元的铁死亡;同时,在探究ACSL4的表达和胶质瘤细胞的铁死亡与增殖的关系的研究中发现ACSL4的过表达会降低GPX4的表达,并增加铁质化标志物(5-羟基二十四碳烯(HETE)、12-HETE和 15-HETE)的水平,从而促进胶质瘤细胞的铁死亡;此外,ACSL4的过表达会降低细胞活力,使细胞存活率下降。)为了进一步证实PKCβII–ACSL4–铁死亡通路与癌症免疫治疗之间的功能联系,作者用载体对照或PKCβII质粒稳定转染B16-PKCβII敲除细胞。植入小鼠体内后,用PD1抗体、Lipro-1或者两者一起来治疗小鼠。正如预期的那样,系统性PD1抗体靶向治疗显著提高了脂质过氧化以及活化的细胞毒性CD4+T和CD8+T细胞水平,并抑制了过表达PKCβII的B16-PKCβII-/-移殖小鼠模型体内的肿瘤生长。但在B16-PKCβII敲除肿瘤异种移植的小鼠模型体内并没有检测到抑制肿瘤生长的效应(附图10a–g)。此外,在重新过表达PKCβII的肿瘤中,铁死亡抑制剂Lipro-1显著抑制了PD1介导的抗肿瘤作用(附图10)。因此,PKCβII通过促进铁死亡提高了免疫治疗的效果,Lipro-1显著抑制了基于PD1治疗介导的抗肿瘤作用。
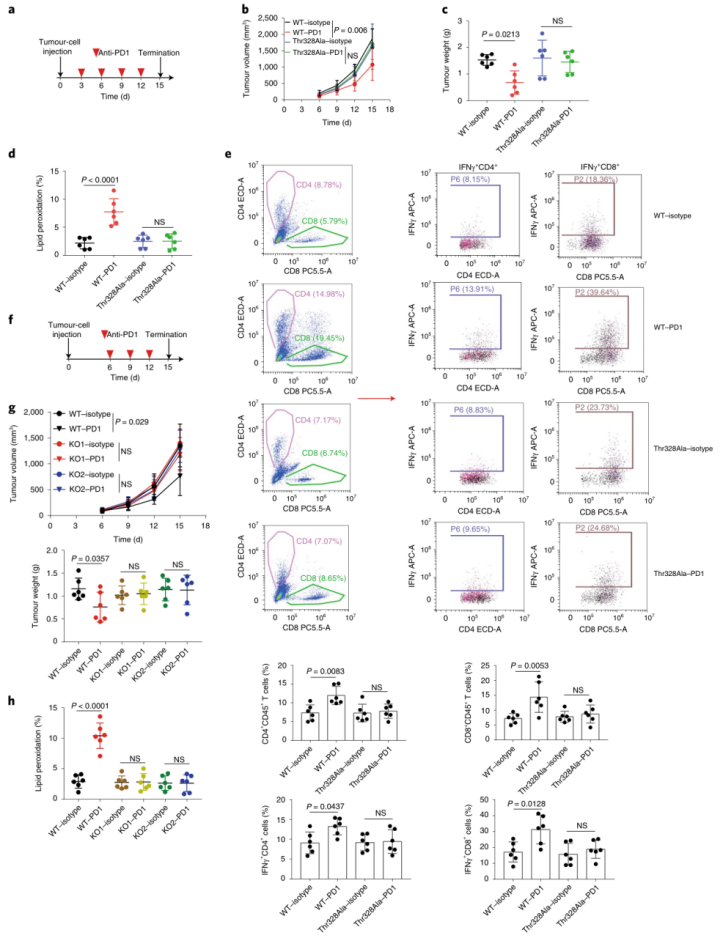
图5 PKCβII–ACSL4通路的减弱通过抑制铁死亡而降低免疫治疗的效果
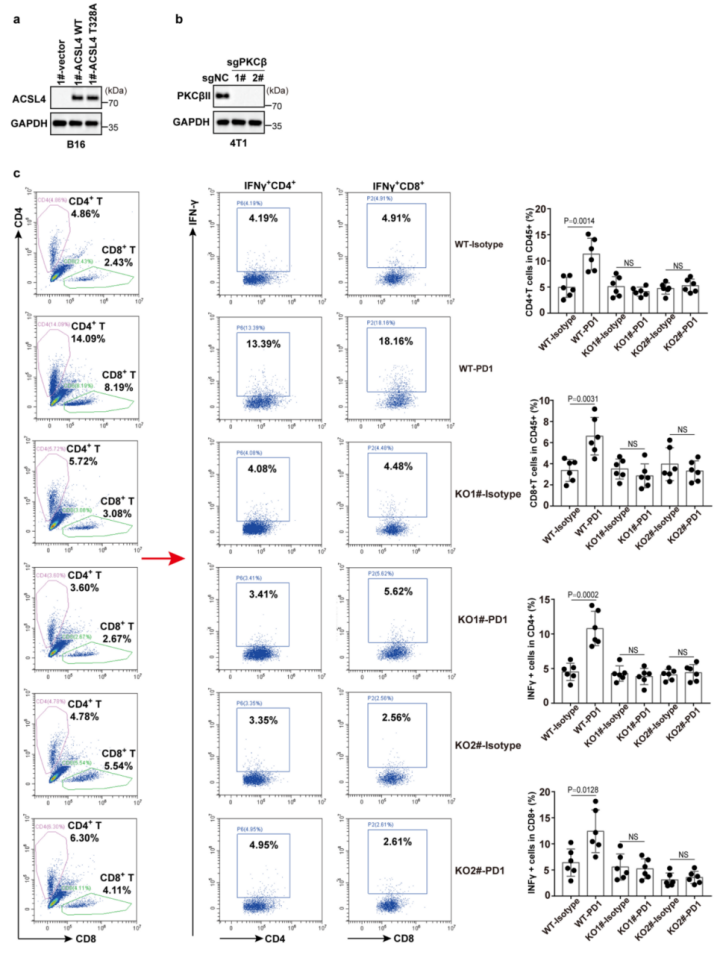
附图9
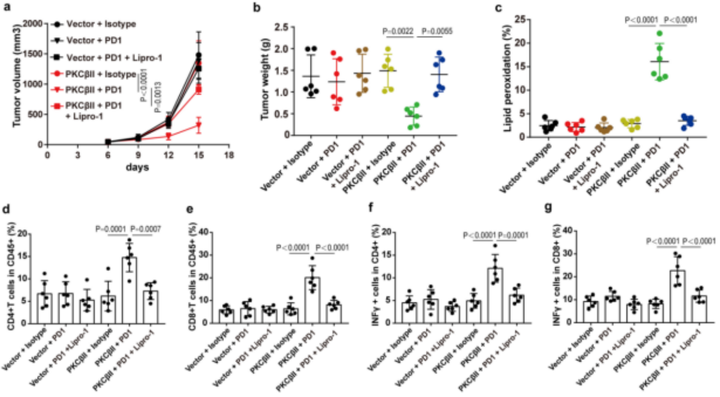
附图10
总结
在本篇研究中,作者发现PKCβII磷酸化ACSL4能够增强脂质过氧化进而诱导铁死亡。首先,作者通过独立的全基因组CRISPR–Cas9和激酶抑制剂文库筛选结果发现“PKCβII激活是铁死亡的关键”。为阐明PKCβII诱导肿瘤细胞铁死亡的分子机制,作者利用免疫共沉淀、磷酸化位点预测、体外激酶、特异性磷酸化抗体制备及脂质组学等方法证实,PKCβII磷酸化ACSL4T328位点,促进ACSL4激活。激活的ACSL4显著促进含不饱和脂肪酸磷脂的合成,进一步诱导脂质过氧化物的产生。这样,脂质过氧化-PKCβII-ACSL4正反馈轴的持续运转,启动了脂质过氧化的快速扩增过程,最终诱导铁死亡的发生。作者在铁死亡相关的动物模型中进一步证实PKCβII磷酸化ACSL4迅速扩增脂质过氧化的过程在铁死亡中的作用。发现与ACSL4-WT相比,ACSL4-T328A(磷酸化失活突变)肿瘤对PD-1抗体敏感性下降。同样,敲除PKCβII显著抑制肿瘤对PD-1抗体的敏感性,过表达PKCβII能回复肿瘤的免疫治疗疗效,且铁死亡抑制剂显著抑制PKCβII介导的免疫治疗疗效增强。这表明PKCβII-ACSL4通过促进铁死亡增强免疫治疗疗效,提示PKCβII及ACSL4可作为肿瘤免疫治疗疗效的分子标记物和靶标。
本文证实了PKCβII–ACSL4机制能够促进脂质过氧化物的积累来促进铁死亡,且PKCβII–ACSL4轴的减弱能抑制铁死亡,进而削弱癌症免疫治疗的效果,该研究为铁死亡相关的癌症治疗提供了潜在的靶点和策略。
原文链接:https://www.nature.com/articles/s41556-021-00818-3
关注微信公众号代谢学人
了解更多前沿资讯

https://blog.sciencenet.cn/blog-3483272-1332941.html
上一篇:代谢学人--Nature Metabolism 2月刊代谢精选
下一篇:代谢学人--Nature Metabolism 3月刊代谢精选