博文
Compartment Saddle Plot分析细节
||
cooltools saddle命令:
Usage: cooltools saddle [OPTIONS] COOL_PATH TRACK_PATH EXPECTED_PATH
具体运行如下:
cooltools saddle --contact-type cis --strength --out-prefix sample --qrange 0.02 0.98 --fig pdf sample_100000.cool sample_100000.eigs.cis.vecs.tsv sample_100000.exp.tsv
注意:这里cool必须用balance之后的
计算步骤如下:
1. 通过cooler.Cooler()载入cool文件 => 赋于变量clr
2. 通过pandas.read_table载入compartment eigs文件(即sample_100000.eigs.cis.vecs.tsv,在程序中这个文件称为track_path),默认读入的列为chrom,start,end,E1 => 赋于变量track
3. 通过make_cooler_view函数(内部调用bioframe.make_viewframe)从cool文件中拿到chromsize。如果提供的--view选项(指定查看的区域),则会仅取这段区域,否则用全部区域来做compartment saddle plot => view_df
4. 对track按染色体分组,之后每组中start最小值和end最大值构建dataframe => track_view_df
5. 结合view_df信息载入expected文件(即sample_100000.exp.tsv,在程序中称为expected_path),载入时内部调用pandas.read_table。默认读入的列为balanced.avg
options:
如果指定--min-dist,当这个值小于0时,min_diag为3,否则为ceil(指定的长度/binsize);同样如果指定--max_dist,当这个值大于0时floor(指定的长度/binsize)
6. 对track做mask,具体将weight为空值的bin,对应的E1也设为空值,在整个过程中还会多次出现mask空值
7. 通过align_track_with_cooler函数,这一步是做track的预处理
8. 关键步骤
利用api.saddle.digitize对track进行digitize。即先按vrange和qrange取这列值的大于某个值和小于某个值的区间内的值,再按n_bins对这些值进行分档。
具体代码如下:
digitized[digitized_col] = np.digitize(track_values, binedges, right=False)
测试发现binedges为单调递增的,所以binedges为0的bins属于compartment B,binedges最大的bins属于compartment A

digitize操作可用以下例子理解:
某次考试有100道题,一题1分,这样考试结果为0-100分,为了将这100个分为5个档次,人为的设定0-20分为E档,20-40分为D档,...,80-100分A档。这样每个考生的成绩都被成为A-E 5个档中的一个,这个过程就是digitize。其中均分为5个档,对应n_bins=5
digitize示意图如下:
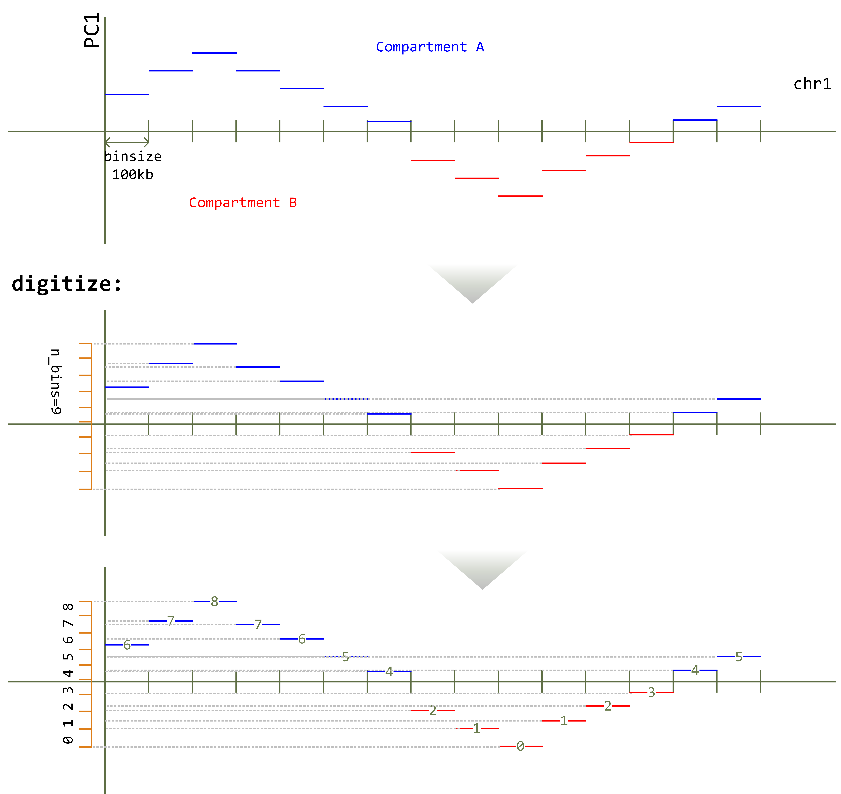
注:输入文件最好所有binsize都一致,可以用100kb、200kb或者400kb,视基因组大小而定
9. 关键步骤
采用api.saddle.saddle先转OE矩阵,再对同一个档的互作频率取平均
9.1 转OE矩阵
它的核心步骤先采用_make_cis_obsexp_fetcher(如果是cis)或者_make_trans_obsexp_fetcher(如果是trans)获取OE矩阵,它整合cool matrix和expected结果,获取observed/expected结果,具体如下:
以"chr16"的cis互作为例,reg1_coords和reg2_coords均为('chr16', 0, 53896199),即(染色体名,染色体起始位点,染色体终止位点)
reg1_coords = tuple(view_df.loc[reg1])
# reg1="chr16", reg1_coords=('chr16', 0, 53896199)
reg2_coords = tuple(view_df.loc[reg2])
# reg2="chr16", reg2_coords=('chr16', 0, 53896199)
observed matrix通过以下方式获得:
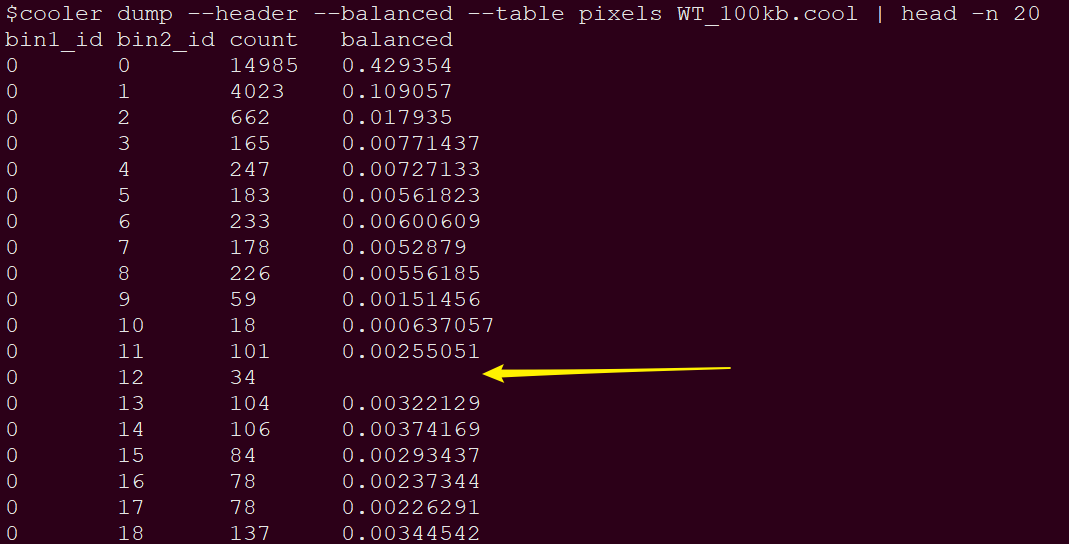
obs_mat = clr.matrix(balance=clr_weight_name).fetch(reg1_coords, reg2_coords)
这里observed matrix是balance之后的矩阵,可以通过以下命令看到,balanced值中会有一些空值,这些在程序中也是有处理的
expected matrix通过以下方式获得:
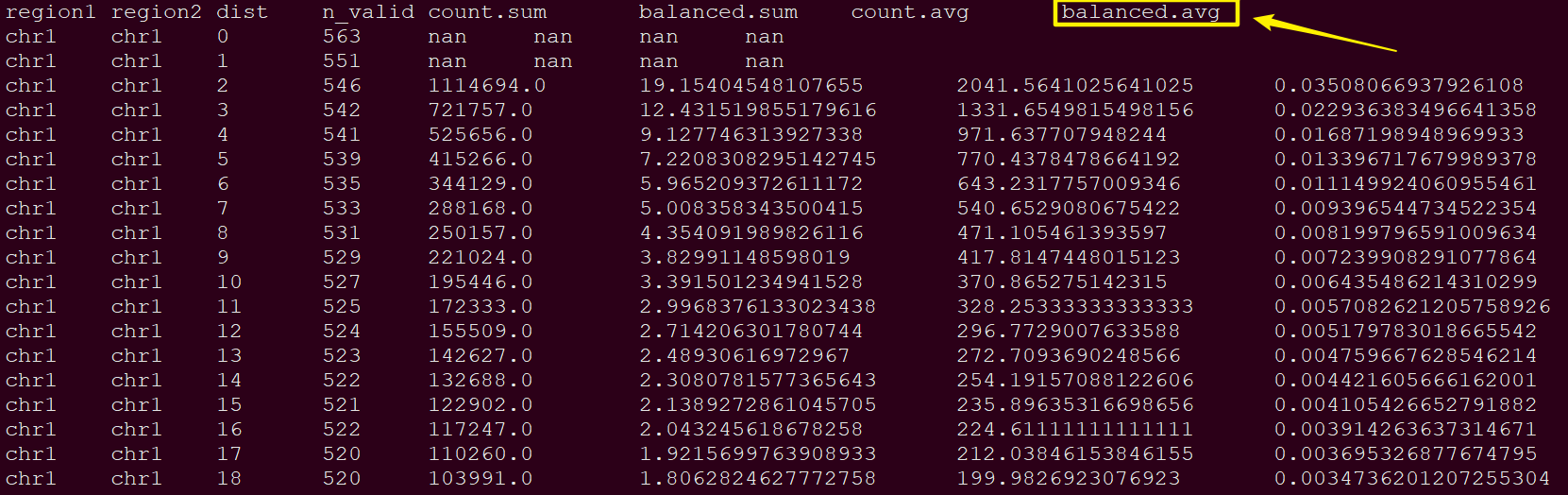
exp_mat = toeplitz(expected[reg1, reg2][: obs_mat.shape[0]])
expected数据格式如下:
{('chr16', 'chr16'): array([nan, nan, 0.03315045, ..., 0.00093574, 0.00171282, ...]), ("chr17", "chr17"): array([nan, nan, 0.03460911, ..., 0.00219353, 0.00286887, ...]), ...}
array中的值为expected文件balanced.avg列的值
toeplitz是from scipy.linalg import toeplitz载入的,该矩阵描述如下:托普利兹矩阵,简称为T型矩阵,托普利兹矩阵的主对角线上的元素相等,平行于主对角线的线上的元素也相等;矩阵中的各元素关于次对角线对称,即T型矩阵为次对称矩阵。以下是wiki上的解释:

再获取OE矩阵,即obs_mat / exp_mat
以后也可以利用这种方法来获取单染色体OE矩阵,进而从其中提取出每个区段上的子矩阵
生成OE矩阵的方法如下示意图:

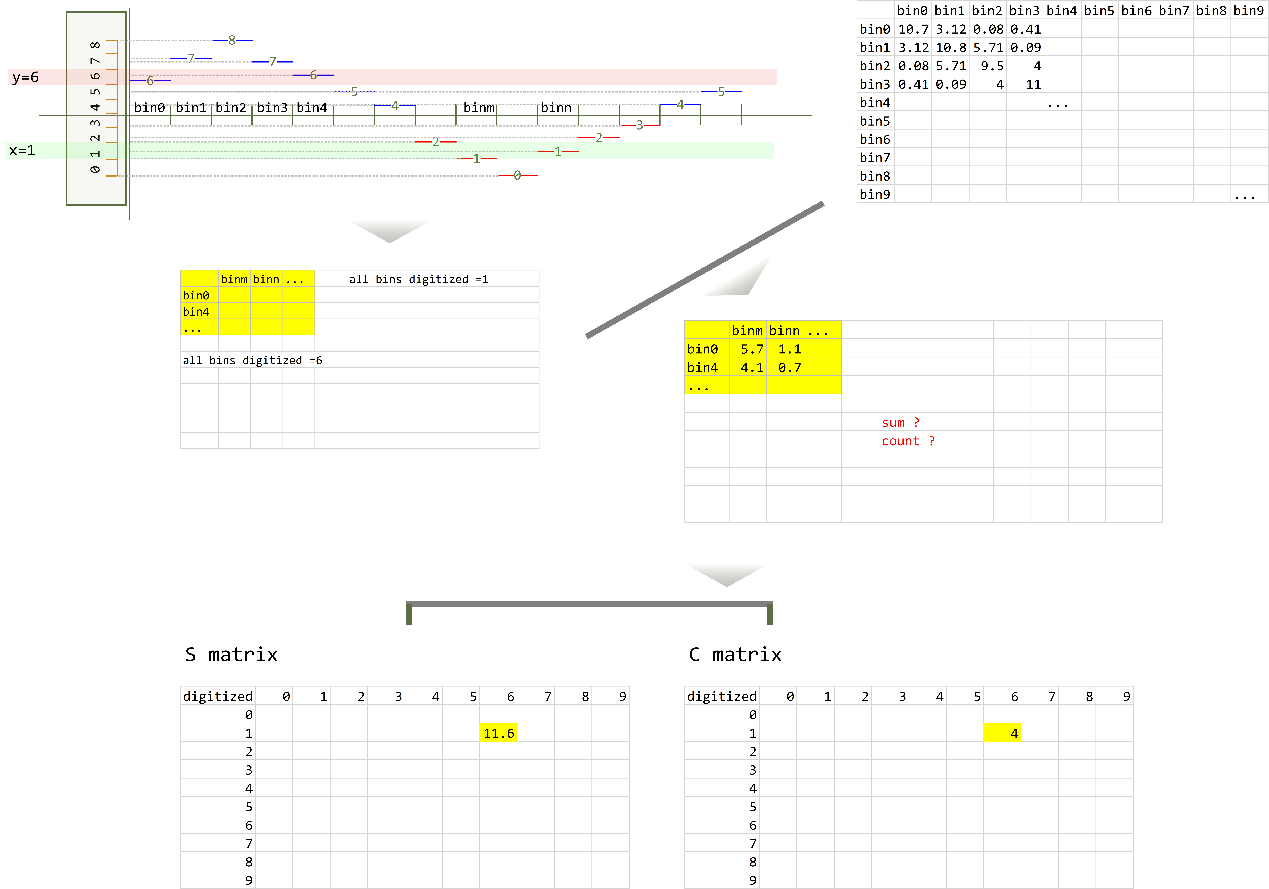
9.2 采用_accumulate函数对PC1在横纵坐标同一个档的元素做sum和数目计数,具体作法如下:
for i in range(n_bins): # bin数目循环,对应x轴
row_mask = digitized[reg1] == i
# PC1 digitized列表中,分档后的值为i的那些行
for j in range(n_bins): # bin数目循环,对应y轴
col_mask = digitized[reg2] == j
# PC1 digitized列表中,分档后的值为j的那些行
data = matrix[row_mask, :][:, col_mask]
# 取出这些xy同一档的元素在OE矩阵中的值
data = data[np.isfinite(data)]
# 去掉这些结果中OE矩阵对应的值为空的那些
S[i, j] += np.sum(data)
# accumulate的S矩阵为这些元素的值的和
C[i, j] += float(len(data))
# accumulate的C矩阵为这些元素的总个数(count)
这一步非常巧妙!因为i和j都是从小到大来的,所以矩阵的[0, 0],即左上角是最小值对应BB (这也是做np.digitize时binedges大小顺序与PC1一致导致的),同时矩阵也是按照位置进行了排序,所以矩阵右下角是最大值AA,同时因为OE matrix是toeplitz矩阵,即沿对角线对称,所以最后拿到的矩阵也是沿对角线对称的,即AB和BA是对称的
有了S和C两个矩阵就可以将两者相除,获得平均值,写入saddledata:
saddledata = S / C
accumulate过程的示意图如下:
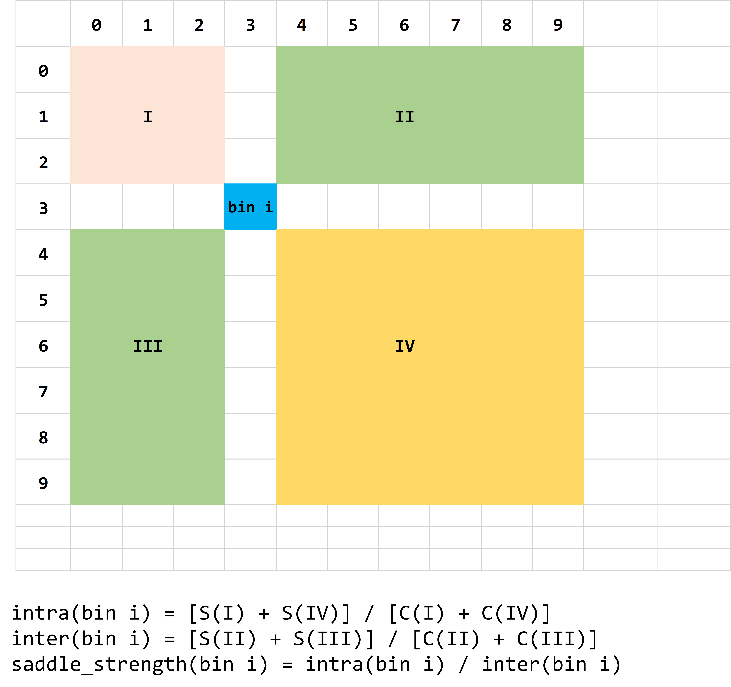
10. 计算saddle strength
如果指定了--strength选项,会调用api.saddle.saddle_strength()函数,它以S和C作为输入,在输出saddledata的同时,还会输出saddle_strength,这是一列数值,它的计算方法是沿着每个bin滑动,以这个bin左上角的正方形加上右下角的正方形里的sum值之和,除以这两个正方形区域的count值之和,得到所谓intra值;右上角和左下角的矩形框的sum除以这两个区域总count,得到inter值,saddle_strength = intra / inter。
所以,如果想标出saddle plot中的strength,其实只需要找到前后20%的两个bins对应的这个值即可,即如果n_bins=50,则找到第10个和第40个bin对应的saddle strength值标记出来即可
saddle strength计算方法如下示意图:
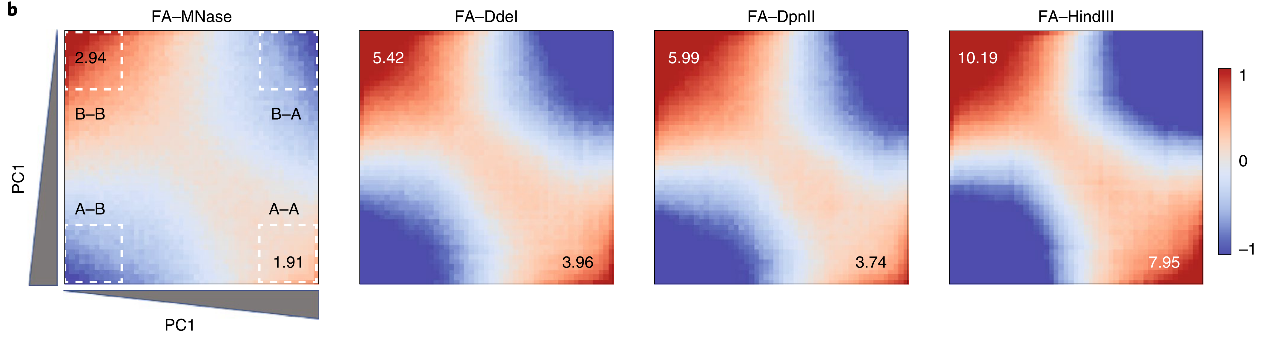
另外,也有一些文章中将AA放到左上角,BB放在右下角,这可能是经过调整的。我们采用cooltools命令行计算的saddle plot,BB和AA的位置如HiC3.0引文(Betul Akgol Oksuz, 2021)中的描述一致:
the upper left corner of the interaction matrix represents the strongest B-B interactions, the lower right represents the strongest A-A interactions, and the upper right and lower left represent B-A and A-B, respectively.
参考材料:
[1] Betul Akgol Oksuz, Liyan Yang, Sameer Abraham, et al. Systematic evaluation of chromosome conformation capture assays. 2021. Nature methods
[2] https://github.com/open2c/cooltools
https://blog.sciencenet.cn/blog-2970729-1367103.html
上一篇:pybedtools filter bug 避坑
下一篇:HiC-Pro3.1.0安装问题及解决办法